Université Pierre et Marie Curie, UMR 7238, Equipe de Génomique Analytique, Paris, France ; CNRS, UMR 7238, Laboratoire de Génomique des Microorganismes, Paris, France.
PLoS Comput Biol. 2013;9(12):e1003369. doi: 10.1371/journal.pcbi.1003369. Epub 2013 Dec 5.
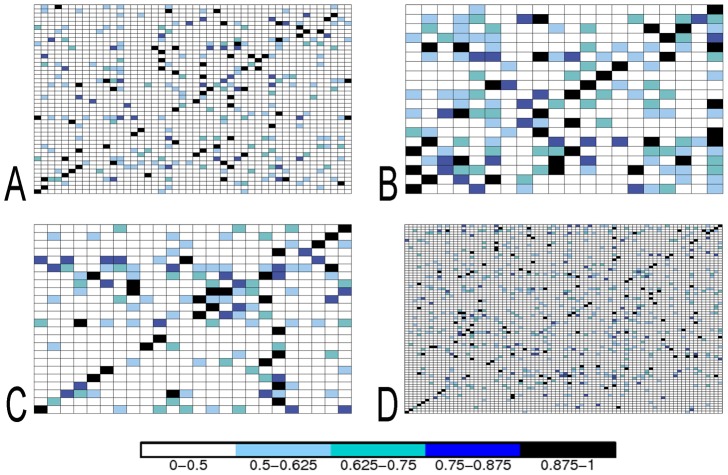
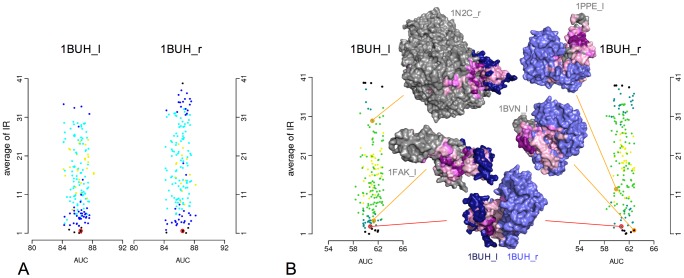
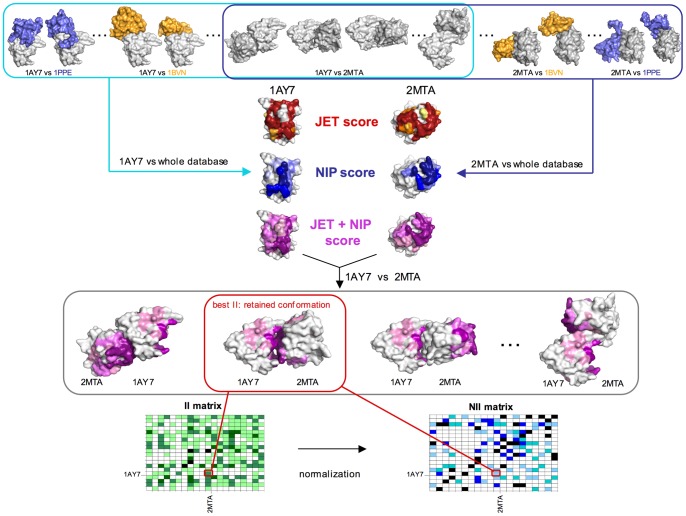
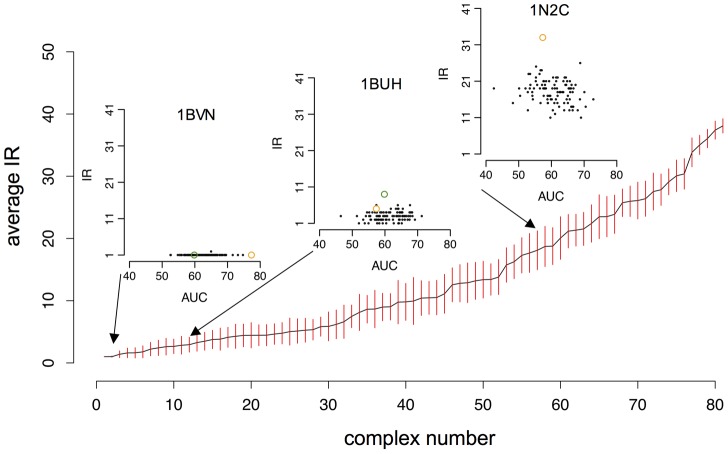
Large-scale analyses of protein-protein interactions based on coarse-grain molecular docking simulations and binding site predictions resulting from evolutionary sequence analysis, are possible and realizable on hundreds of proteins with variate structures and interfaces. We demonstrated this on the 168 proteins of the Mintseris Benchmark 2.0. On the one hand, we evaluated the quality of the interaction signal and the contribution of docking information compared to evolutionary information showing that the combination of the two improves partner identification. On the other hand, since protein interactions usually occur in crowded environments with several competing partners, we realized a thorough analysis of the interactions of proteins with true partners but also with non-partners to evaluate whether proteins in the environment, competing with the true partner, affect its identification. We found three populations of proteins: strongly competing, never competing, and interacting with different levels of strength. Populations and levels of strength are numerically characterized and provide a signature for the behavior of a protein in the crowded environment. We showed that partner identification, to some extent, does not depend on the competing partners present in the environment, that certain biochemical classes of proteins are intrinsically easier to analyze than others, and that small proteins are not more promiscuous than large ones. Our approach brings to light that the knowledge of the binding site can be used to reduce the high computational cost of docking simulations with no consequence in the quality of the results, demonstrating the possibility to apply coarse-grain docking to datasets made of thousands of proteins. Comparison with all available large-scale analyses aimed to partner predictions is realized. We release the complete decoys set issued by coarse-grain docking simulations of both true and false interacting partners, and their evolutionary sequence analysis leading to binding site predictions. Download site: http://www.lgm.upmc.fr/CCDMintseris/
基于粗粒分子对接模拟和进化序列分析得出的结合位点预测,可以对具有多种结构和界面的数百种蛋白质进行大规模的蛋白质-蛋白质相互作用分析。我们在 Mintseris 基准 2.0 的 168 个蛋白质上证明了这一点。一方面,我们评估了相互作用信号的质量以及对接信息与进化信息的贡献,结果表明两种信息的结合可以提高伙伴识别的能力。另一方面,由于蛋白质相互作用通常发生在具有多个竞争伙伴的拥挤环境中,我们对具有真正伙伴的蛋白质的相互作用进行了彻底的分析,也对与非伙伴的相互作用进行了分析,以评估与真正伙伴竞争的环境中的蛋白质是否会影响其识别。我们发现了三种蛋白质群体:强烈竞争、从不竞争和具有不同程度相互作用的群体。这些群体和强度水平用数字进行了描述,并为蛋白质在拥挤环境中的行为提供了特征。我们表明,在某种程度上,伙伴识别并不取决于环境中存在的竞争伙伴,某些生化类别的蛋白质本质上比其他蛋白质更容易分析,而且小蛋白质并不比大蛋白质更杂乱无章。我们的方法揭示了结合位点的知识可以用于降低对接模拟的高计算成本,而不会对结果的质量产生影响,这证明了将粗粒对接应用于由数千个蛋白质组成的数据集的可能性。并与所有可用的旨在进行伙伴预测的大规模分析进行了比较。我们发布了由真正和虚假相互作用伙伴的粗粒对接模拟产生的完整诱饵集,以及它们的进化序列分析导致的结合位点预测。下载网址:http://www.lgm.upmc.fr/CCDMintseris/