Singh Anshika N, Sharma Neeti
Symbiosis School of Biological Sciences, Symbiosis International University, Gram - Lavale, Taluka - Mulshi, Pune, India.
Onco Targets Ther. 2017 Oct 10;10:4925-4933. doi: 10.2147/OTT.S144725. eCollection 2017.
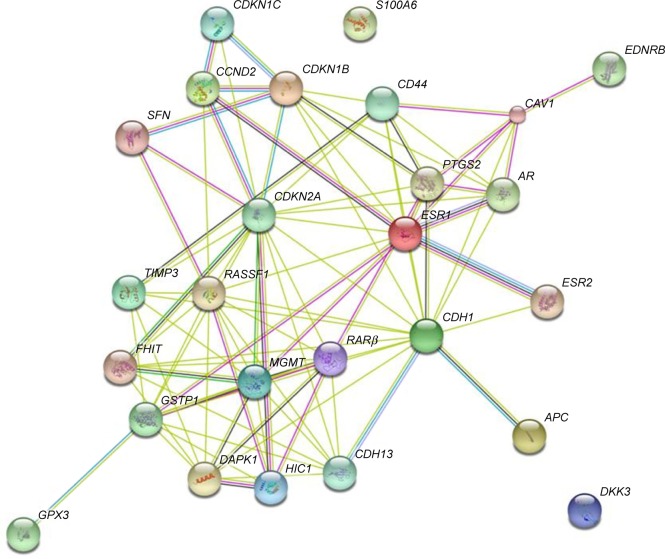
Prostate cancer (PCa), a multifocal clinically heterogeneous disease, is the most commonly diagnosed non-cutaneous neoplasm in men worldwide. The epigenome of PCa is a typical representation of catastrophic model of epigenetic alterations during tumorigenesis and its progression. Alterations in methylation patterns in tumor suppressors, cell cycle, oncogenes and metabolism-related genes are the most commonly observed epigenetic alterations in PCa. In this study, we have developed a computational strategy to identify methylated biomarker signature panels as potential targets of PCa by screening >160 genes reported to be epigenetically dysregulated, and shortlisted 26 differentially methylated genes (DMGs) that significantly contribute to oncogenesis. The gene ontology and functional enrichment analysis were performed, which showed that identified DMGs contribute to cellular and metabolic processes such as cell communication, cell cycle, response to drugs, apoptosis and p53 signaling. The top hub genes , , , , , and identified from protein-protein interaction network construction using Search Tool for the Retrieval of Interacting Genes contributed to hormonal response, inflammatory response, cell cycle, reactive oxygen species activity and receptor kinase activity, which are related to hallmarks of cancer as revealed by their functional enrichment analysis by Cytoscape. These genes were further scrutinized for CpG islands, transcription start sites and positions of methylated cytosines to study their methylation profiles. Our analysis revealed high negative correlation values between methylation frequencies and gene expressions of the hub genes, namely, , , , , , and , which can be used as potential diagnostic biomarkers for PCa.
前列腺癌(PCa)是一种多灶性临床异质性疾病,是全球男性中最常见的非皮肤肿瘤。PCa的表观基因组是肿瘤发生及其进展过程中表观遗传改变的灾难性模型的典型代表。肿瘤抑制基因、细胞周期、癌基因和代谢相关基因的甲基化模式改变是PCa中最常见的表观遗传改变。在本研究中,我们开发了一种计算策略,通过筛选超过160个据报道表观遗传失调的基因,来识别甲基化生物标志物特征面板作为PCa的潜在靶点,并筛选出26个对肿瘤发生有显著贡献的差异甲基化基因(DMGs)。进行了基因本体和功能富集分析,结果表明所识别的DMGs参与细胞和代谢过程,如细胞通讯、细胞周期、药物反应、细胞凋亡和p53信号传导。使用基因相互作用检索搜索工具从蛋白质-蛋白质相互作用网络构建中识别出的前几个枢纽基因, 、 、 、 、 、 和 ,参与激素反应、炎症反应、细胞周期、活性氧物种活性和受体激酶活性,这些与癌症特征相关,这在Cytoscape对其进行的功能富集分析中得到了揭示。进一步对这些基因的CpG岛、转录起始位点和甲基化胞嘧啶的位置进行了仔细研究,以研究它们的甲基化谱。我们的分析揭示了枢纽基因 、 、 、 、 、 和 的甲基化频率与基因表达之间的高度负相关值,这些值可作为PCa的潜在诊断生物标志物。