Channing Division of Network Medicine, Brigham and Women's Hospital and Harvard Medical School, Boston, MA, 02115, USA.
Science and Technology on Information Systems Engineering Laboratory, National University of Defense Technology, Changsha, Hunan, 410073, China.
Nat Commun. 2017 Dec 11;8(1):2042. doi: 10.1038/s41467-017-02090-2.
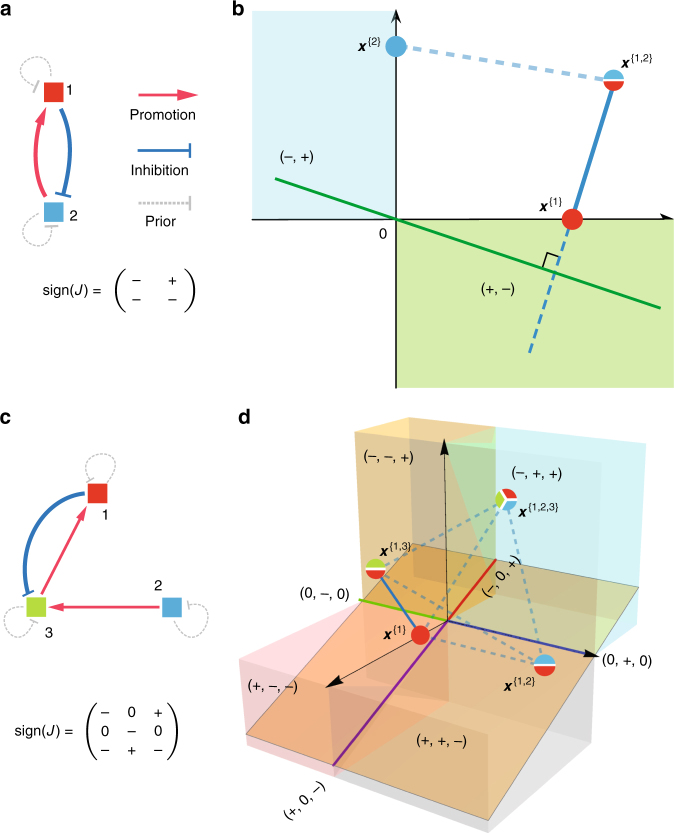
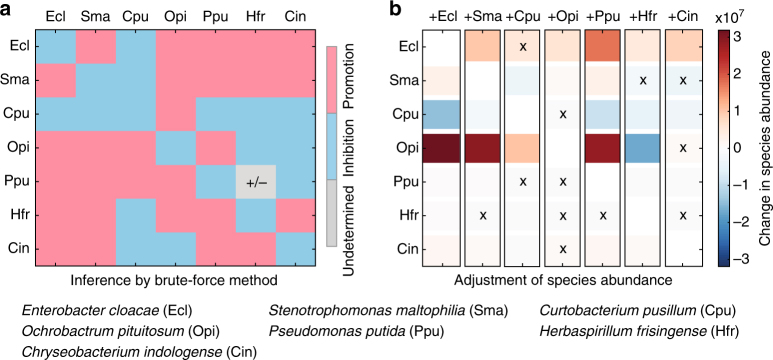
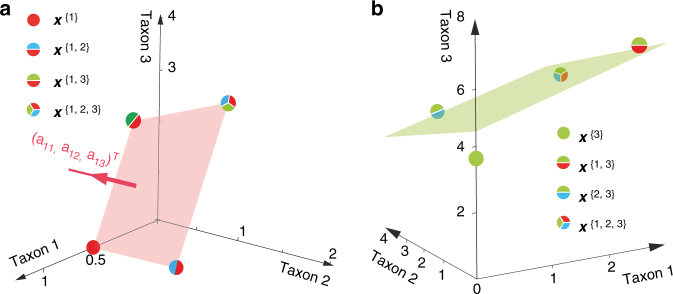
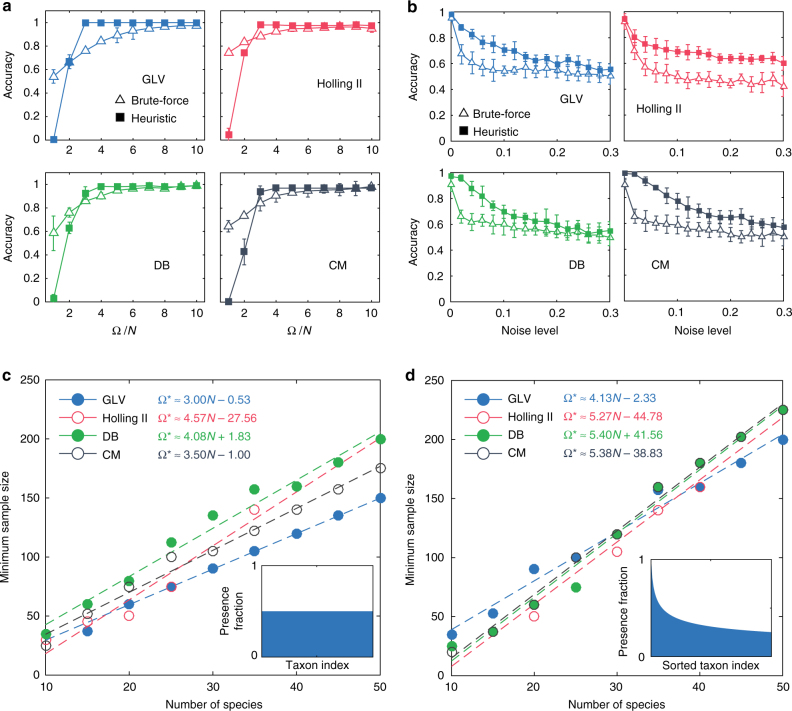
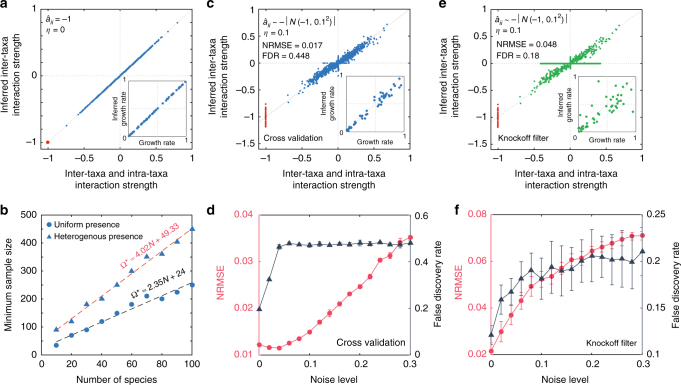
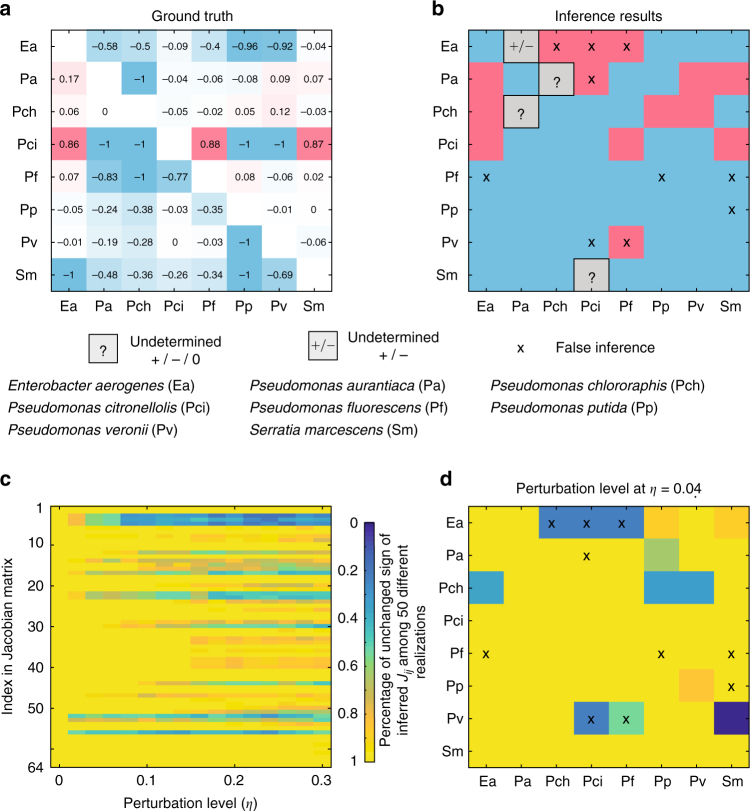
Mapping the ecological networks of microbial communities is a necessary step toward understanding their assembly rules and predicting their temporal behavior. However, existing methods require assuming a particular population dynamics model, which is not known a priori. Moreover, those methods require fitting longitudinal abundance data, which are often not informative enough for reliable inference. To overcome these limitations, here we develop a new method based on steady-state abundance data. Our method can infer the network topology and inter-taxa interaction types without assuming any particular population dynamics model. Additionally, when the population dynamics is assumed to follow the classic Generalized Lotka-Volterra model, our method can infer the inter-taxa interaction strengths and intrinsic growth rates. We systematically validate our method using simulated data, and then apply it to four experimental data sets. Our method represents a key step towards reliable modeling of complex, real-world microbial communities, such as the human gut microbiota.
绘制微生物群落的生态网络是理解其组装规则和预测其时间行为的必要步骤。然而,现有的方法需要假设特定的种群动态模型,而该模型是事先未知的。此外,这些方法需要拟合纵向丰度数据,而这些数据通常不足以进行可靠的推断。为了克服这些限制,我们在这里开发了一种基于稳态丰度数据的新方法。我们的方法可以在不假设任何特定种群动态模型的情况下推断网络拓扑和种间相互作用类型。此外,当种群动态被假设为遵循经典的广义Lotka-Volterra 模型时,我们的方法可以推断种间相互作用强度和内在增长率。我们使用模拟数据系统地验证了我们的方法,然后将其应用于四个实验数据集。我们的方法代表了朝着可靠建模复杂真实世界微生物群落(如人类肠道微生物群)迈出的关键一步。