Department of Cell and Molecular Biology, Science for Life Laboratory, Karolinska Institutet, PO Box 285, SE-171 77, Stockholm, Sweden.
Stockholm Bioinformatics Center, Department of Biochemistry and Biophysics, Stockholm University, Science for Life Laboratory, Box 1031, SE-171 21, Solna, Sweden.
Sci Rep. 2018 Jan 8;8(1):28. doi: 10.1038/s41598-017-18341-7.
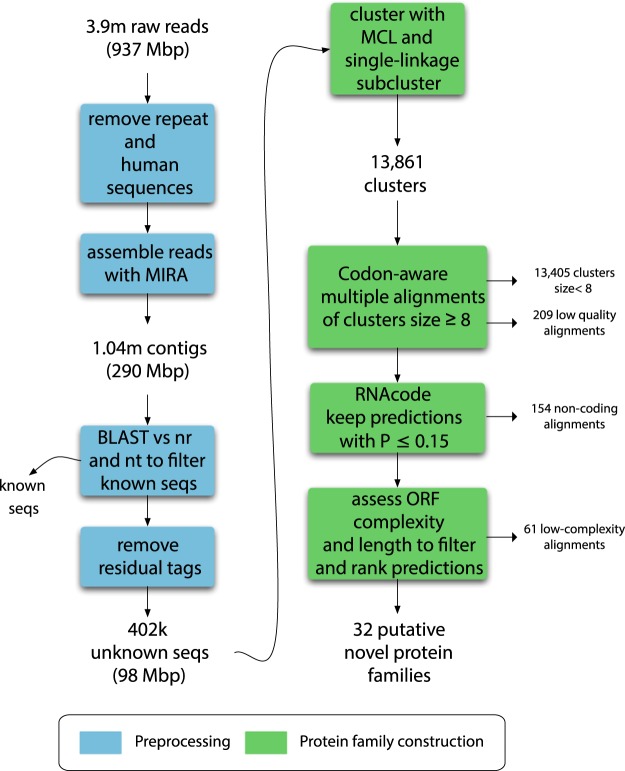
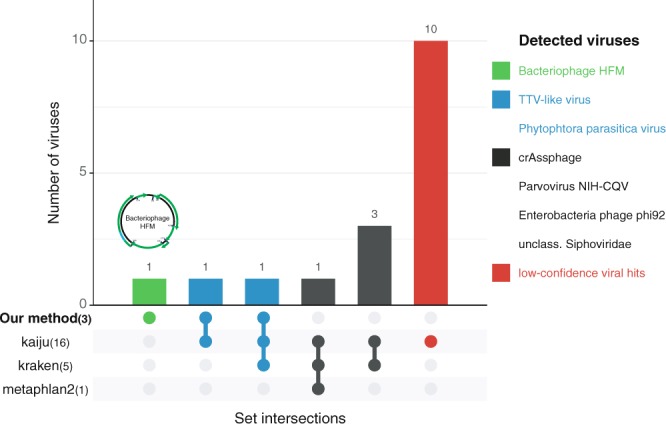
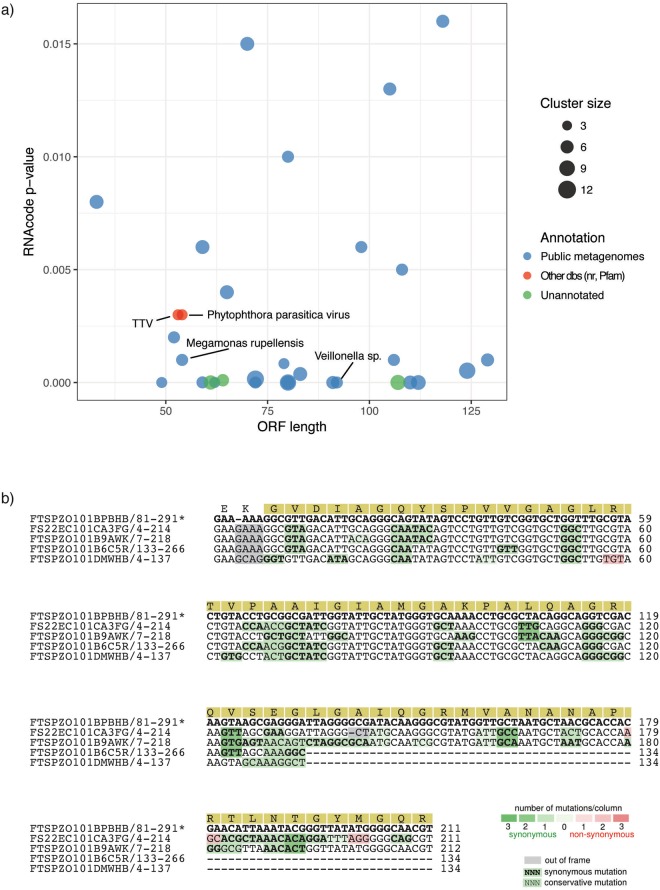
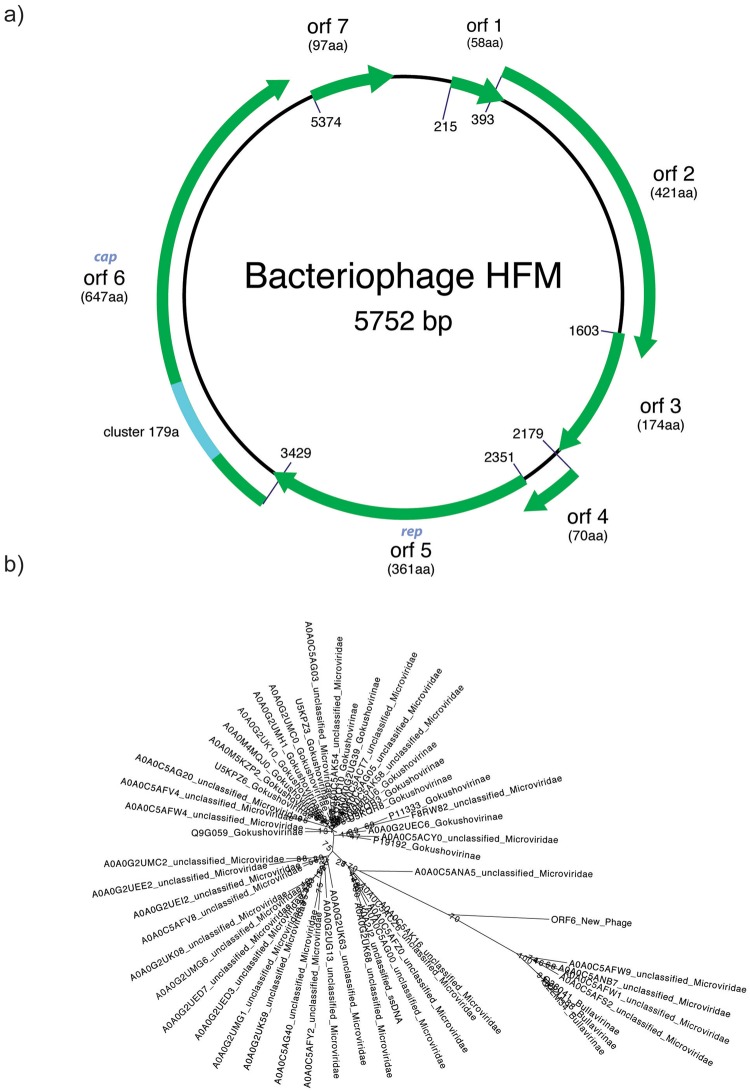
Massive amounts of metagenomics data are currently being produced, and in all such projects a sizeable fraction of the resulting data shows no or little homology to known sequences. It is likely that this fraction contains novel viruses, but identification is challenging since they frequently lack homology to known viruses. To overcome this problem, we developed a strategy to detect ORFan protein families in shotgun metagenomics data, using similarity-based clustering and a set of filters to extract bona fide protein families. We applied this method to 17 virus-enriched libraries originating from human nasopharyngeal aspirates, serum, feces, and cerebrospinal fluid samples. This resulted in 32 predicted putative novel gene families. Some families showed detectable homology to sequences in metagenomics datasets and protein databases after reannotation. Notably, one predicted family matches an ORF from the highly variable Torque Teno virus (TTV). Furthermore, follow-up from a predicted ORFan resulted in the complete reconstruction of a novel circular genome. Its organisation suggests that it most likely corresponds to a novel bacteriophage in the microviridae family, hence it was named bacteriophage HFM.
目前正在产生大量的宏基因组学数据,在所有这些项目中,相当一部分的结果数据与已知序列没有或几乎没有同源性。这部分数据很可能包含新的病毒,但由于它们通常与已知病毒没有同源性,因此鉴定具有挑战性。为了克服这个问题,我们开发了一种在鸟枪法宏基因组学数据中检测 ORFan 蛋白家族的策略,使用基于相似性的聚类和一组过滤器来提取真正的蛋白家族。我们将这种方法应用于 17 个源自人鼻咽抽吸物、血清、粪便和脑脊液样本的病毒富集文库。这导致了 32 个预测的假定新基因家族。一些家族在重新注释后显示出与宏基因组数据集和蛋白质数据库中的序列有可检测的同源性。值得注意的是,一个预测的家族与高度可变的 Torque Teno 病毒 (TTV) 的一个 ORF 相匹配。此外,对一个预测的 ORFan 的后续研究导致了一个新的圆形基因组的完整重建。它的组织表明,它很可能对应于微病毒科中的一种新型噬菌体,因此它被命名为噬菌体 HFM。