Malaghan Institute of Medical Research, Wellington, New Zealand.
Department of Biological Sciences, King Abdulaziz University, Jeddah, Saudi Arabia.
BMC Biol. 2018 Jan 11;16(1):6. doi: 10.1186/s12915-017-0473-4.
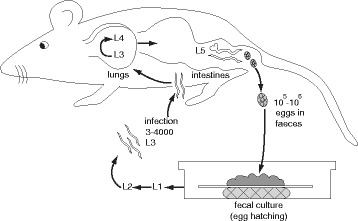
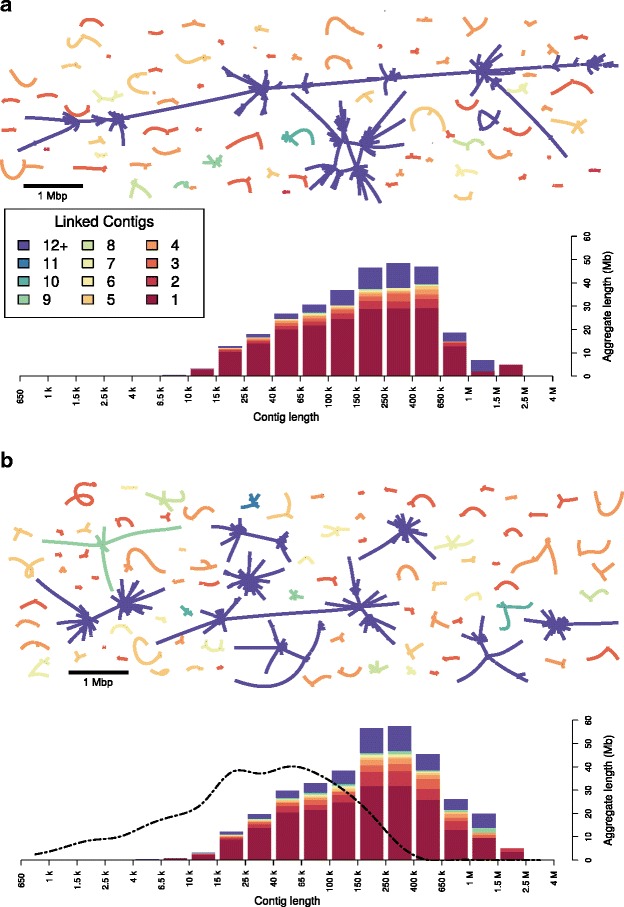
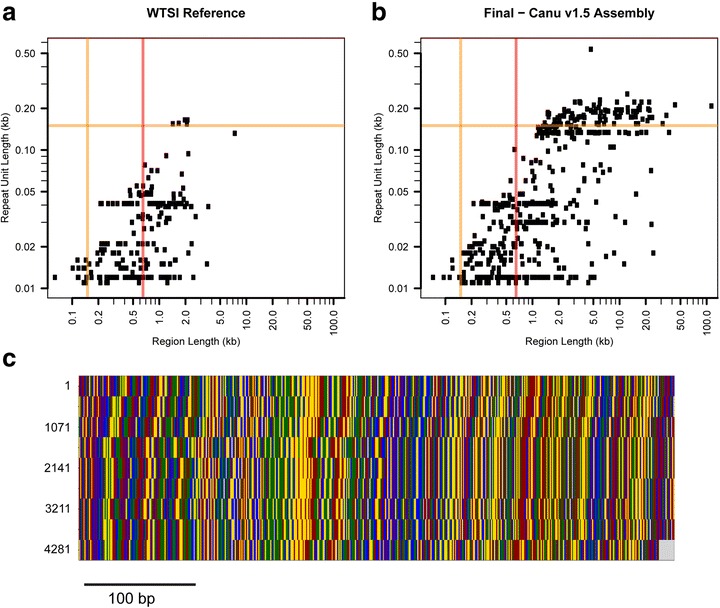
Eukaryotic genome assembly remains a challenge in part due to the prevalence of complex DNA repeats. This is a particularly acute problem for holocentric nematodes because of the large number of satellite DNA sequences found throughout their genomes. These have been recalcitrant to most genome sequencing methods. At the same time, many nematodes are parasites and some represent a serious threat to human health. There is a pressing need for better molecular characterization of animal and plant parasitic nematodes. The advent of long-read DNA sequencing methods offers the promise of resolving complex genomes.
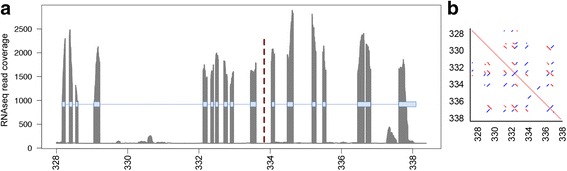
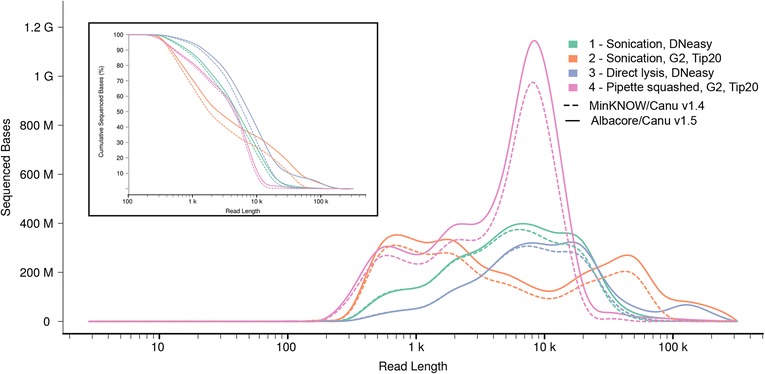
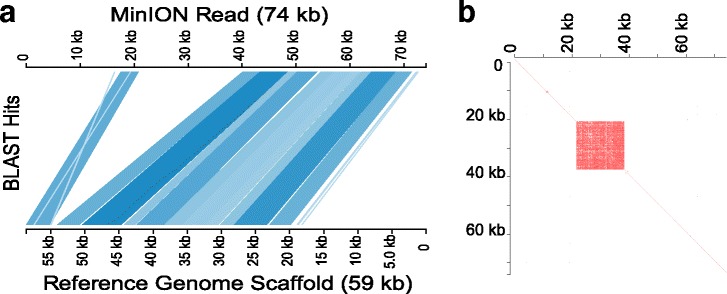
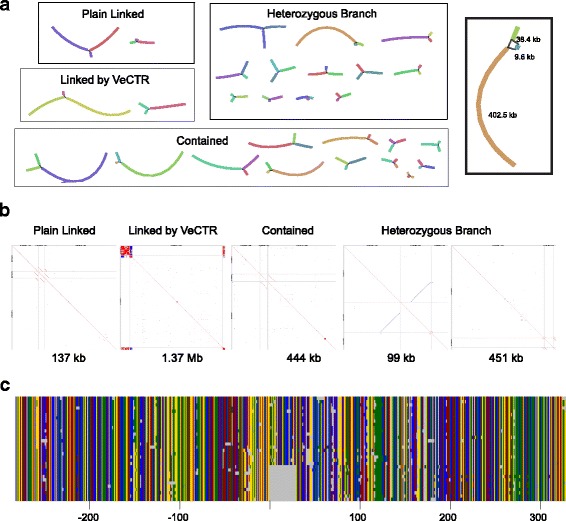
Using Nippostrongylus brasiliensis as a test case, applying improved base-calling algorithms and assembly methods, we demonstrate the feasibility of de novo genome assembly matching current community standards using only MinION long reads. In doing so, we uncovered an unexpected diversity of very long and complex DNA sequences repeated throughout the N. brasiliensis genome, including massive tandem repeats of tRNA genes.
Base-calling and assembly methods have improved sufficiently that de novo genome assembly of large complex genomes is possible using only long reads. The method has the added advantage of preserving haplotypic variants and so has the potential to be used in population analyses.
真核生物基因组组装仍然是一个挑战,部分原因是复杂的 DNA 重复序列普遍存在。对于全染色体线虫来说,这是一个特别严重的问题,因为它们的基因组中存在大量的卫星 DNA 序列。这些序列对大多数基因组测序方法都具有抗性。与此同时,许多线虫是寄生虫,有些对人类健康构成严重威胁。因此迫切需要更好地对动植物寄生线虫进行分子特征描述。长读长 DNA 测序方法的出现为解决复杂基因组提供了希望。
我们以巴西牛带绦虫为研究对象,应用改进的碱基调用算法和组装方法,证明了仅使用 MinION 长读长即可实现符合当前社区标准的从头基因组组装的可行性。在这样做的过程中,我们发现了巴西牛带绦虫基因组中存在大量意想不到的非常长且复杂的 DNA 序列,包括 tRNA 基因的大规模串联重复。
碱基调用和组装方法已经得到了足够的改进,仅使用长读长即可实现大型复杂基因组的从头组装。该方法还有一个额外的优势,即可以保留单倍型变体,因此有可能用于群体分析。