Department of Neurology and Neurological Science, Graduate School of Medicine, Tokyo Medical and Dental University, Tokyo, Japan.
Department of Neurology, Yokufukai Geriatric Hospital, Tokyo, Japan.
J Diabetes Investig. 2018 Nov;9(6):1239-1254. doi: 10.1111/jdi.12833. Epub 2018 Apr 25.
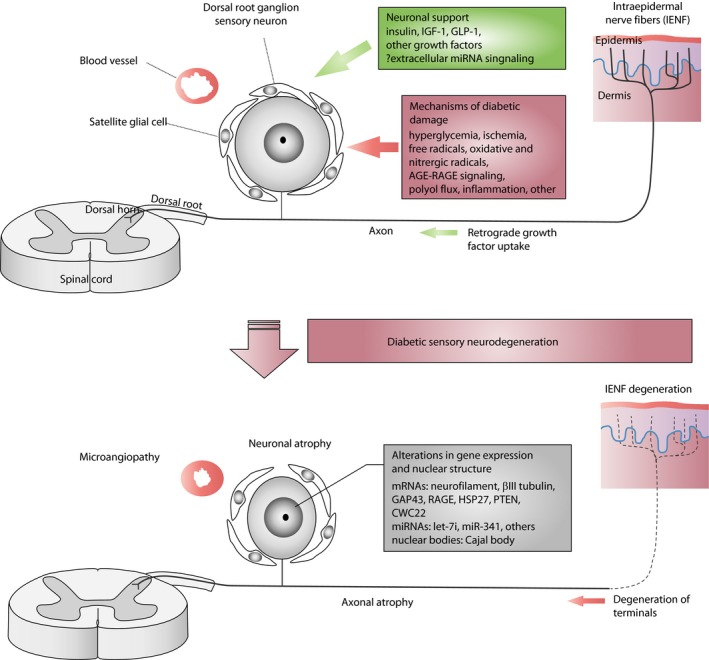
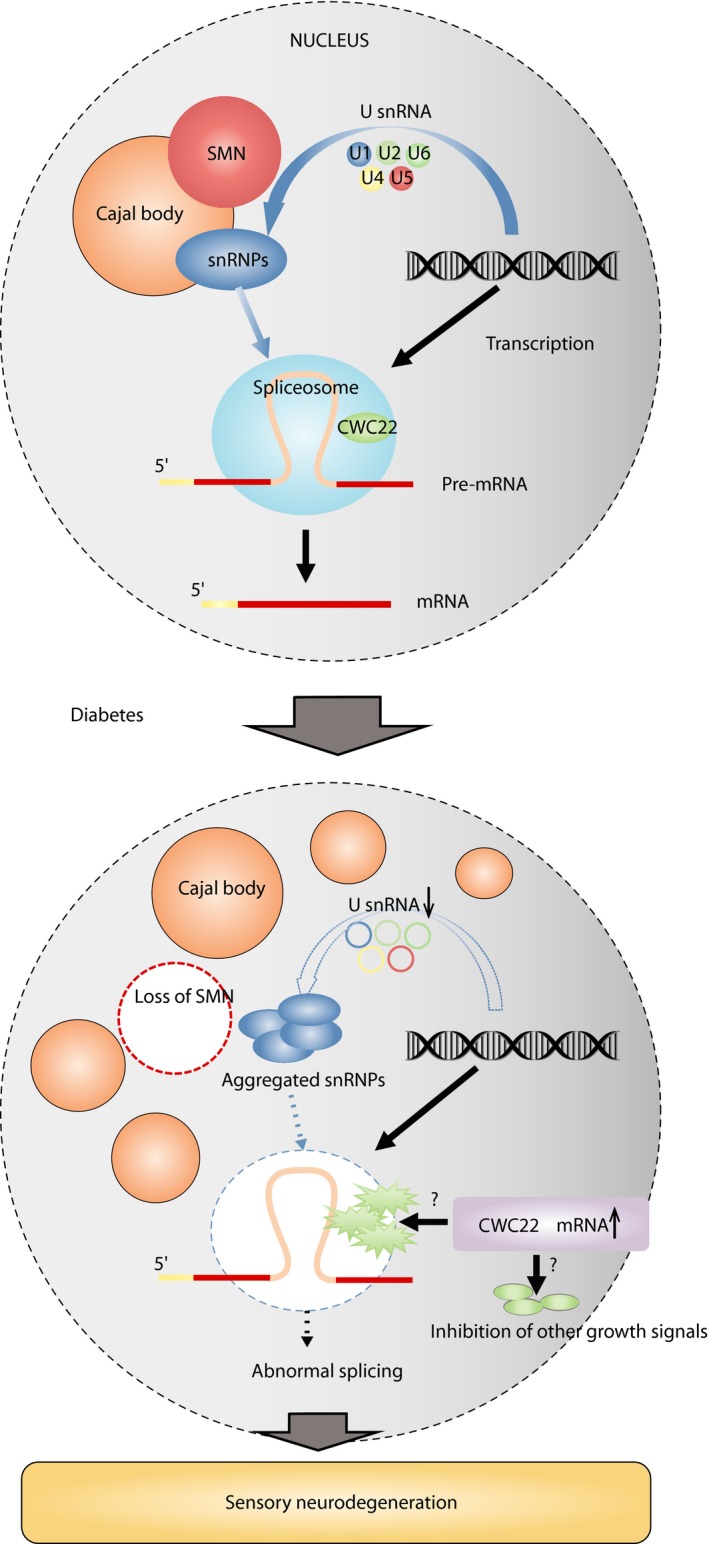
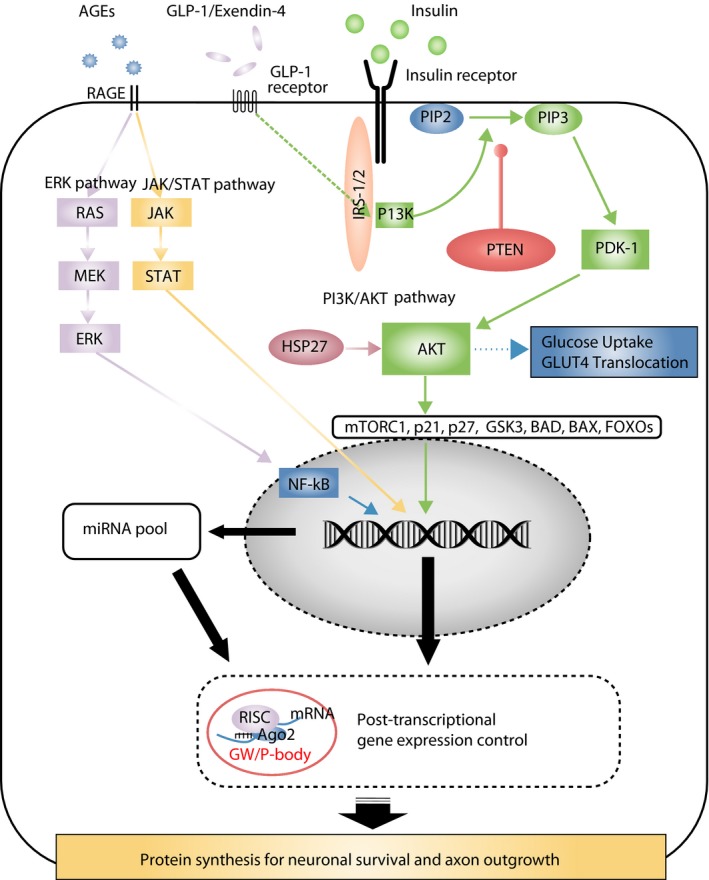
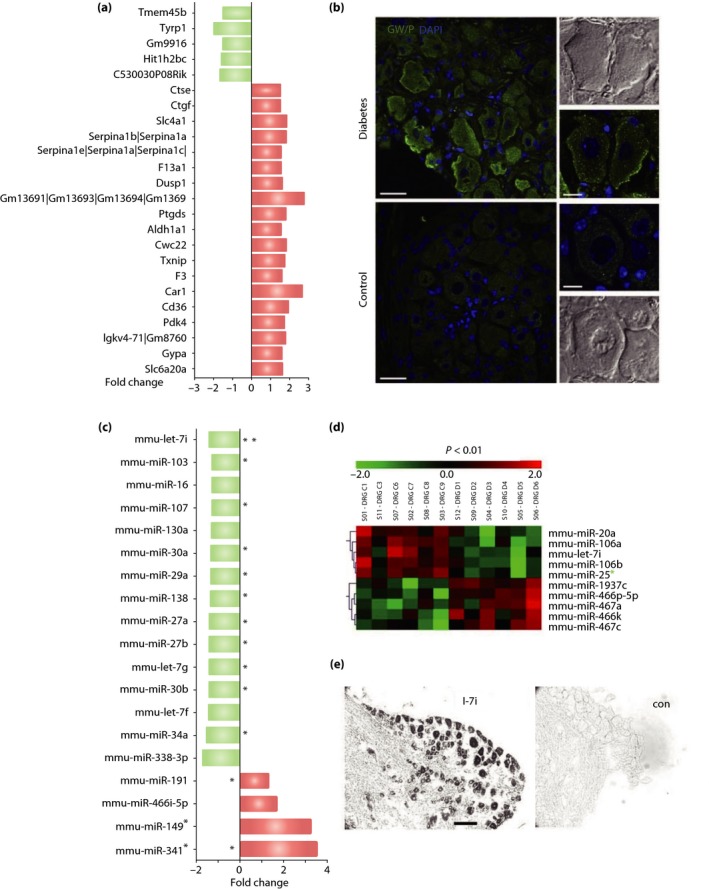
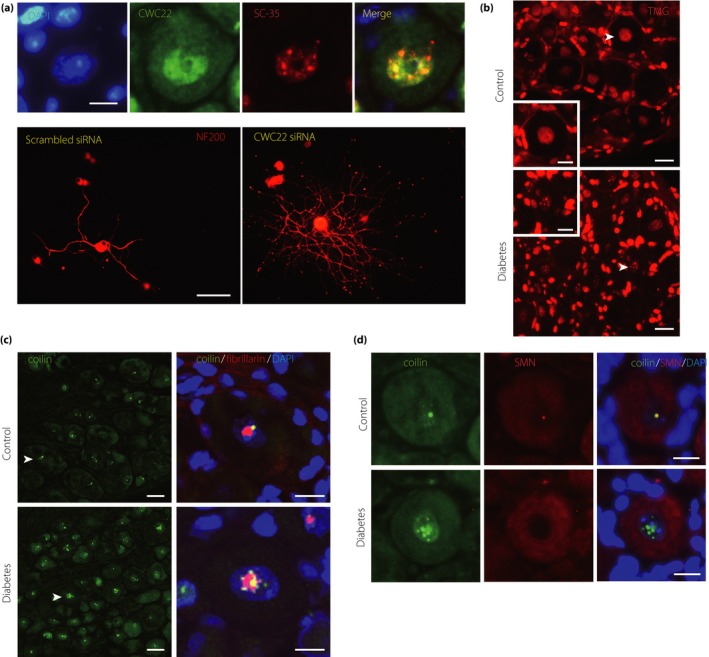
Diabetic polyneuropathy (DPN) continues to be generally considered as a "microvascular" complication of diabetes mellitus alongside nephropathy and retinopathy. The microvascular hypothesis, however, might be tempered by the concept that diabetes directly targets dorsal root ganglion sensory neurons. This neuron-specific concept, supported by accumulating evidence, might account for important features of DPN, such as its early sensory neuron degeneration. Diabetic sensory neurons develop neuronal atrophy alongside a series of messenger ribonucleic acid (RNA) changes related to declines in structural proteins, increases in heat shock protein, increases in the receptor for advanced glycation end-products, declines in growth factor signaling and other changes. Insulin is recognized as a potent neurotrophic factor, and insulin ligation enhances neurite outgrowth through activation of the phosphoinositide 3-kinase-protein kinase B pathway within sensory neurons and attenuates phenotypic features of experimental DPN. Several interventions, including glucagon-like peptide-1 agonism, and phosphatase and tensin homolog inhibition to activate growth signals in sensory neurons, or heat shock protein overexpression, prevent or reverse neuropathic abnormalities in experimental DPN. Diabetic sensory neurons show a unique pattern of microRNA alterations, a key element of messenger RNA silencing. For example, let-7i is widely expressed in sensory neurons, supports their growth and is depleted in experimental DPN; its replenishment improves features of DPN models. Finally, impairment of pre-messenger RNA splicing in diabetic sensory neurons including abnormal nuclear RNA metabolism and structure with loss of survival motor neuron protein, a neuron survival molecule, and overexpression of CWC22, a splicing factor, offer further novel insights. The present review addresses these new aspects of DPN sensory neurodegeneration.
糖尿病性多发性神经病(DPN)通常被认为是糖尿病的一种“微血管”并发症,与肾病和视网膜病变并列。然而,微血管假说可能会受到这样一种概念的影响,即糖尿病直接针对背根神经节感觉神经元。这一神经元特异性概念得到了越来越多的证据的支持,它可能解释了 DPN 的一些重要特征,如早期感觉神经元退化。糖尿病感觉神经元除了一系列与结构蛋白减少、热休克蛋白增加、晚期糖基化终产物受体增加、生长因子信号下降等有关的信使核糖核酸(RNA)变化外,还会发生神经元萎缩。胰岛素被认为是一种有效的神经营养因子,胰岛素结合通过激活感觉神经元中的磷酸肌醇 3-激酶-蛋白激酶 B 通路增强轴突生长,并减轻实验性 DPN 的表型特征。几种干预措施,包括胰高血糖素样肽-1 激动剂和磷酸酶和张力蛋白同源物抑制作用以激活感觉神经元中的生长信号,或热休克蛋白过表达,可预防或逆转实验性 DPN 的神经病变异常。糖尿病感觉神经元显示出独特的 microRNA 改变模式,这是信使 RNA 沉默的关键要素。例如,let-7i 在感觉神经元中广泛表达,支持其生长,并在实验性 DPN 中耗尽;其补充可改善 DPN 模型的特征。最后,糖尿病感觉神经元中前信使 RNA 剪接的损伤,包括异常的核 RNA 代谢和结构,导致运动神经元生存蛋白(一种神经元生存分子)丢失,以及剪接因子 CWC22 的过表达,提供了进一步的新见解。本综述讨论了 DPN 感觉神经退行性变的这些新方面。