Department of Neuroscience, Tufts University School of Medicine, Boston, MA, USA.
Program of Neuroscience, Tufts University Sackler School of Graduate Biomedical Sciences, Boston, MA, USA.
Nature. 2018 Apr;556(7702):505-509. doi: 10.1038/s41586-018-0049-7. Epub 2018 Apr 18.
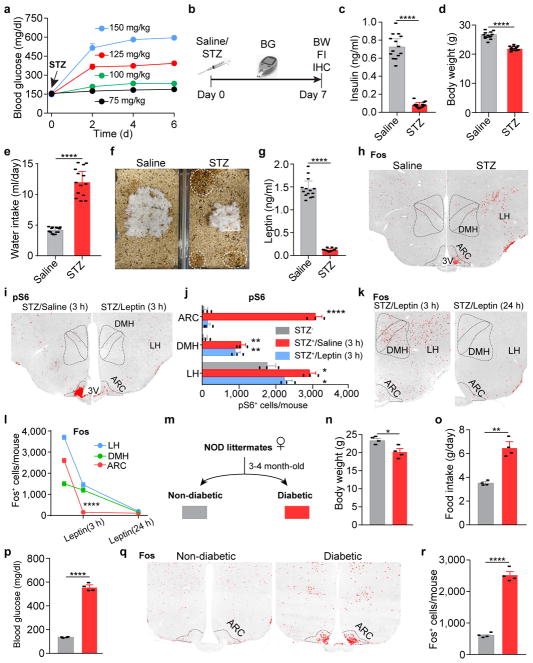
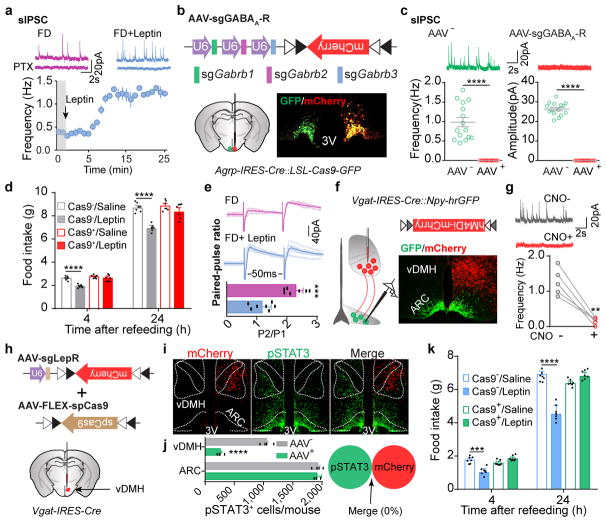
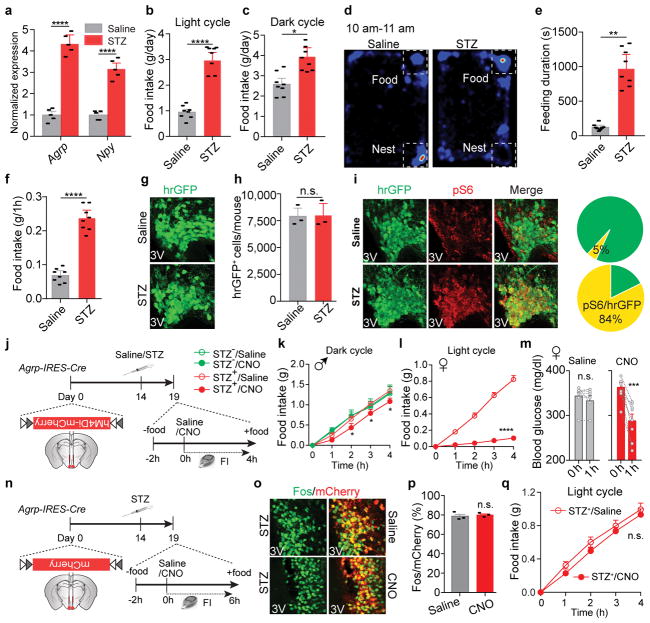
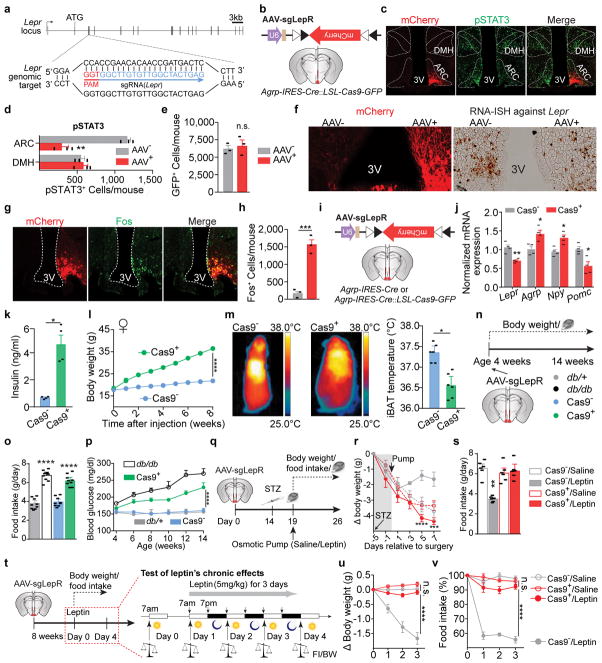
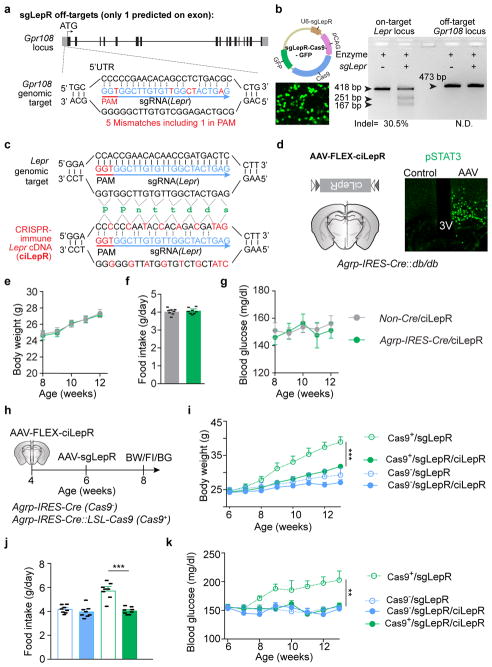
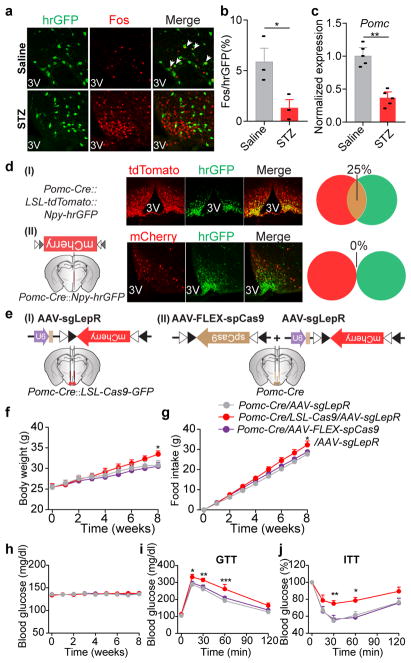
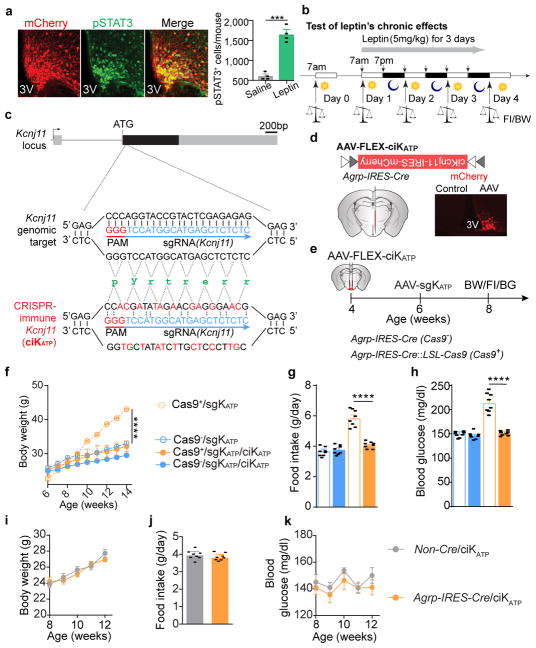
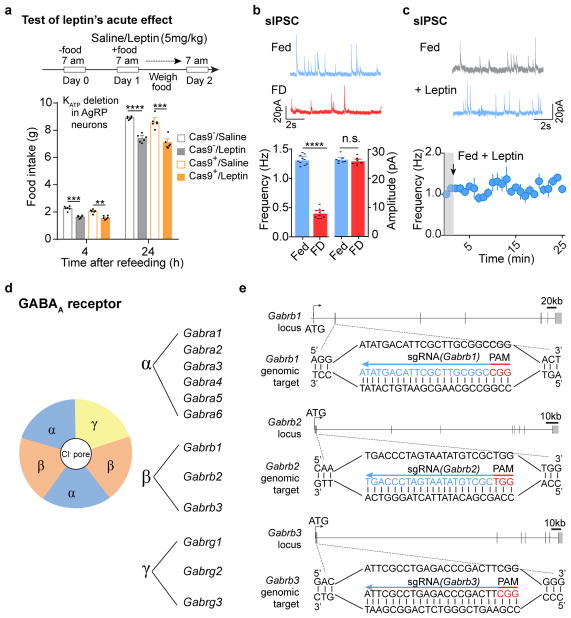
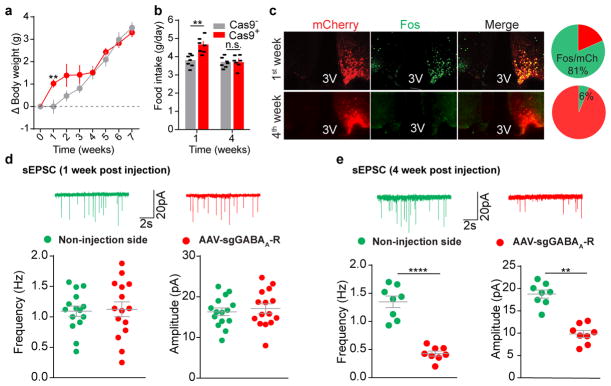
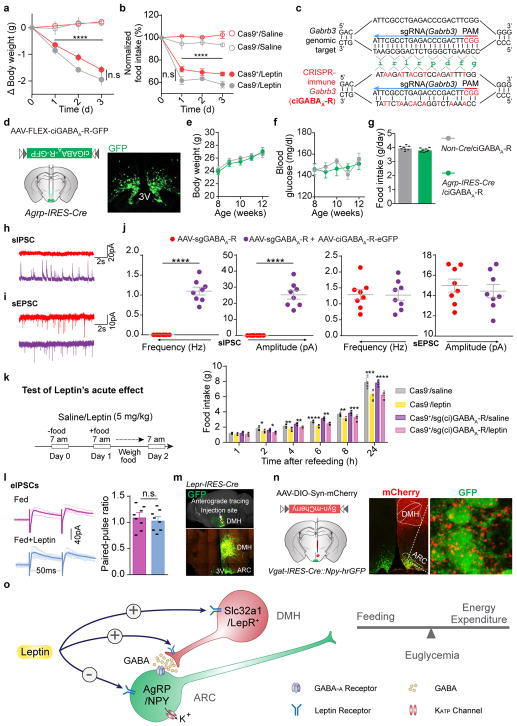
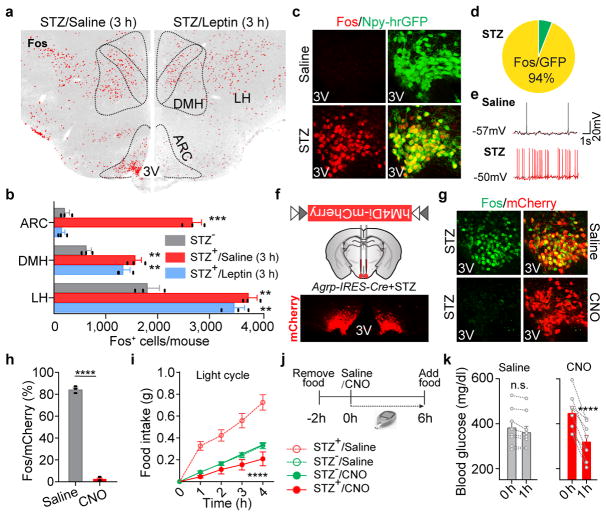
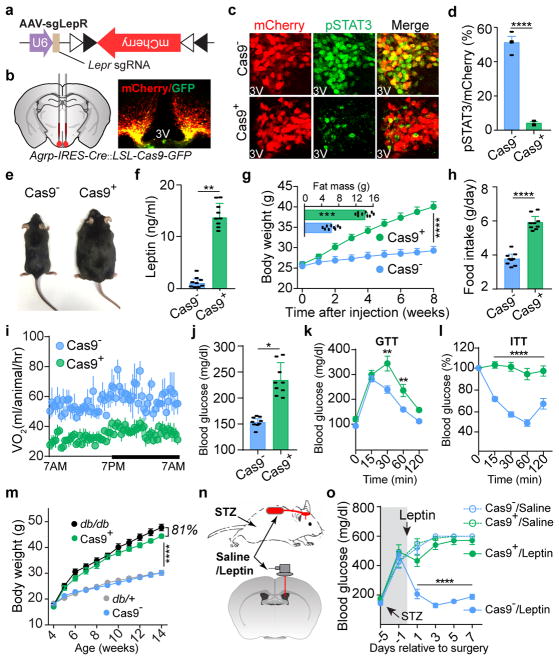
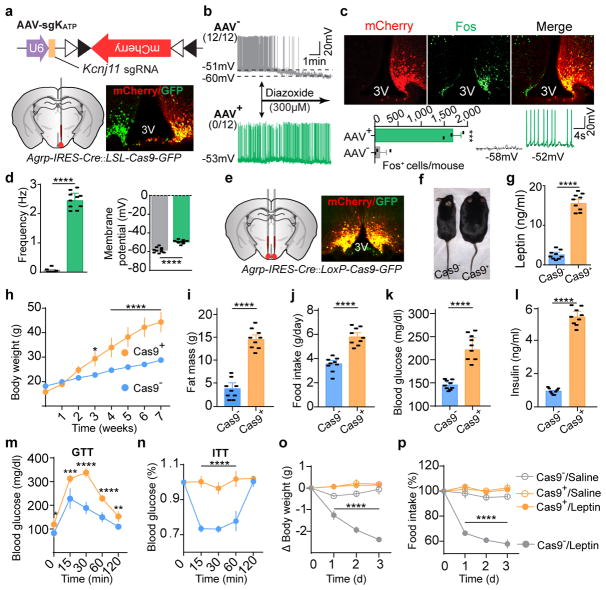
Leptin, a hormone produced in white adipose tissue, acts in the brain to communicate fuel status, suppress appetite following a meal, promote energy expenditure and maintain blood glucose stability. Dysregulation of leptin or its receptors (LEPR) results in severe obesity and diabetes. Although intensive studies on leptin have transformed obesity and diabetes research, clinical applications of the molecule are still limited , at least in part owing to the complexity and our incomplete understanding of the underlying neural circuits. The hypothalamic neurons that express agouti-related peptide (AGRP) and pro-opiomelanocortin (POMC) have been hypothesized to be the main first-order, leptin-responsive neurons. Selective deletion of LEPR in these neurons with the Cre-loxP system, however, has previously failed to recapitulate, or only marginally recapitulated, the obesity and diabetes that are seen in LEPR-deficient Lepr mice, suggesting that AGRP or POMC neurons are not directly required for the effects of leptin in vivo. The primary neural targets of leptin are therefore still unclear. Here we conduct a systematic, unbiased survey of leptin-responsive neurons in streptozotocin-induced diabetic mice and exploit CRISPR-Cas9-mediated genetic ablation of LEPR in vivo. Unexpectedly, we find that AGRP neurons but not POMC neurons are required for the primary action of leptin to regulate both energy balance and glucose homeostasis. Leptin deficiency disinhibits AGRP neurons, and chemogenetic inhibition of these neurons reverses both diabetic hyperphagia and hyperglycaemia. In sharp contrast to previous studies, we show that CRISPR-mediated deletion of LEPR in AGRP neurons causes severe obesity and diabetes, faithfully replicating the phenotype of Lepr mice. We also uncover divergent mechanisms of acute and chronic inhibition of AGRP neurons by leptin (presynaptic potentiation of GABA (γ-aminobutyric acid) neurotransmission and postsynaptic activation of ATP-sensitive potassium channels, respectively). Our findings identify the underlying basis of the neurobiological effects of leptin and associated metabolic disorders.
瘦素是一种在白色脂肪组织中产生的激素,它在大脑中发挥作用,传递燃料状态信息,抑制餐后食欲,促进能量消耗,并维持血糖稳定。瘦素或其受体(LEPR)的失调会导致严重肥胖和糖尿病。尽管对瘦素的深入研究改变了肥胖和糖尿病的研究,但该分子的临床应用仍然有限,至少部分原因是其复杂性和我们对潜在神经回路的不完全理解。表达刺鼠相关肽(AGRP)和前阿黑皮素原(POMC)的下丘脑神经元被假设为主要的一级、瘦素反应神经元。然而,使用 Cre-loxP 系统选择性地删除这些神经元中的 LEPR,以前未能重现或仅轻微重现 Lepr 缺陷型小鼠中所见的肥胖和糖尿病,这表明 AGRP 或 POMC 神经元不是瘦素在体内发挥作用的直接必需条件。因此,瘦素的主要神经靶标仍不清楚。在这里,我们对链脲佐菌素诱导的糖尿病小鼠中的瘦素反应神经元进行了系统、无偏的调查,并利用 CRISPR-Cas9 介导的体内 LEPR 基因消融。出乎意料的是,我们发现 AGRP 神经元而不是 POMC 神经元是瘦素调节能量平衡和葡萄糖稳态的主要作用所必需的。瘦素缺乏会解除 AGRP 神经元的抑制,而这些神经元的化学遗传抑制可逆转糖尿病引起的过度摄食和高血糖。与以前的研究形成鲜明对比的是,我们发现 CRISPR 介导的 AGRP 神经元中的 LEPR 缺失会导致严重肥胖和糖尿病,忠实地复制了 Lepr 小鼠的表型。我们还揭示了瘦素对 AGRP 神经元的急性和慢性抑制的不同机制(分别为 GABA(γ-氨基丁酸)神经传递的突触前增强和 ATP 敏感性钾通道的突触后激活)。我们的研究结果确定了瘦素及其相关代谢紊乱的神经生物学效应的基础。