CERVO Brain Research Center, 2601 Chemin de la Canardière, Québec, Québec, G1J 2G3, Canada.
Department of Psychiatry and Neuroscience, Faculty of Medicine, Université Laval, Québec City, G1V 0A6, Canada.
Neurotherapeutics. 2018 Jul;15(3):715-727. doi: 10.1007/s13311-018-0634-3.
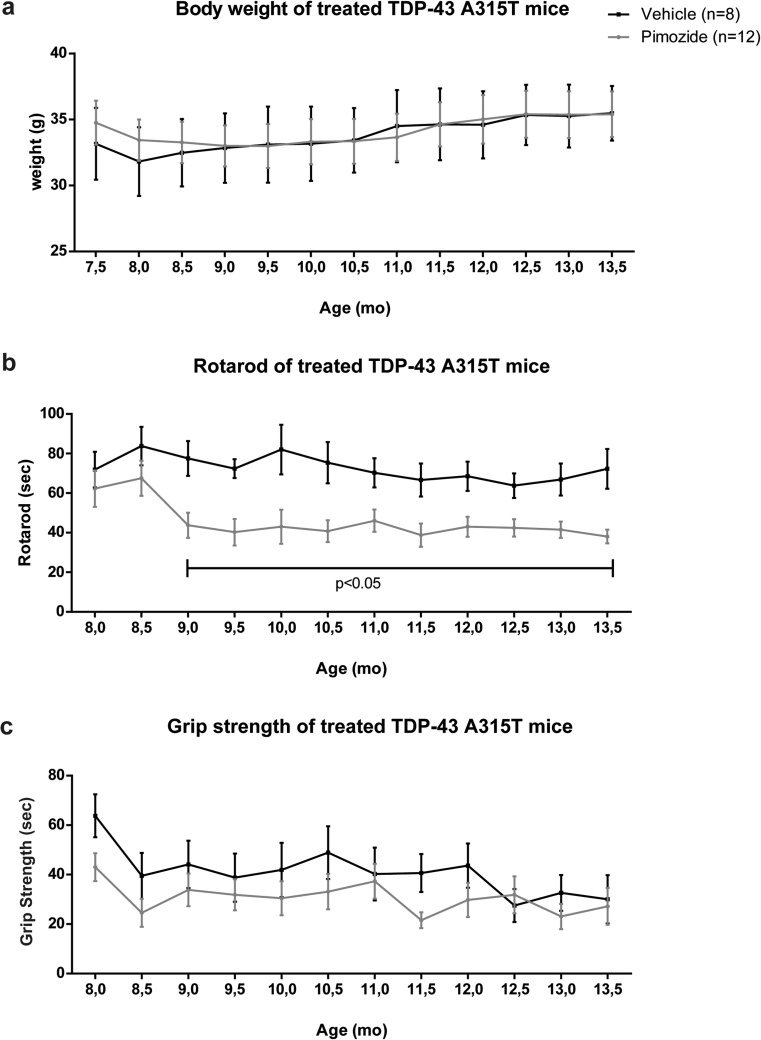
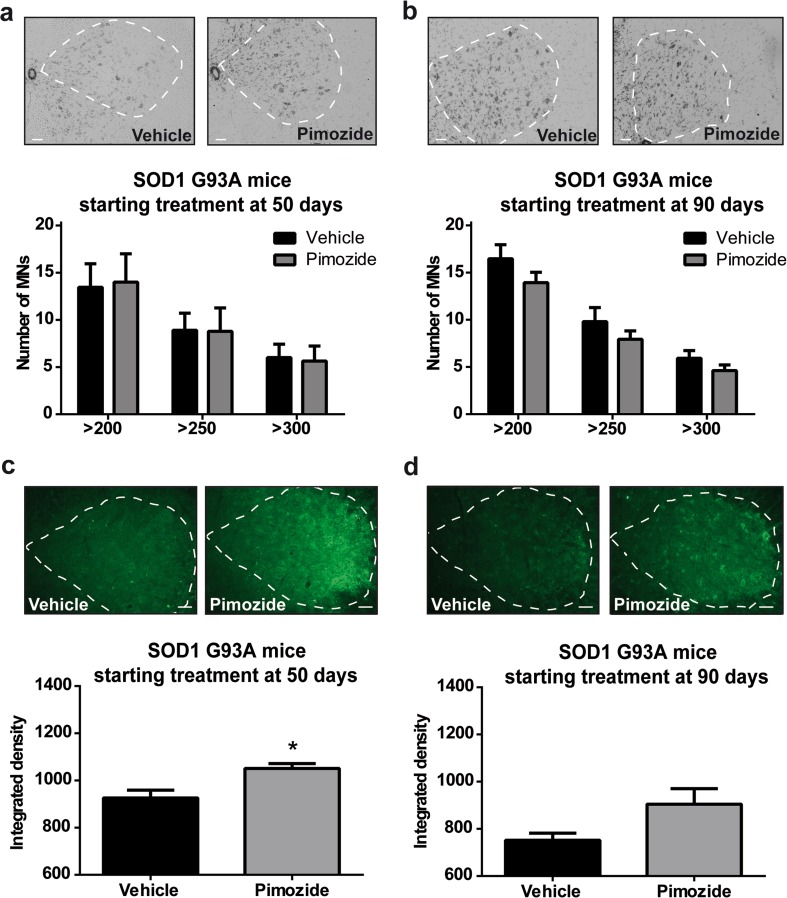
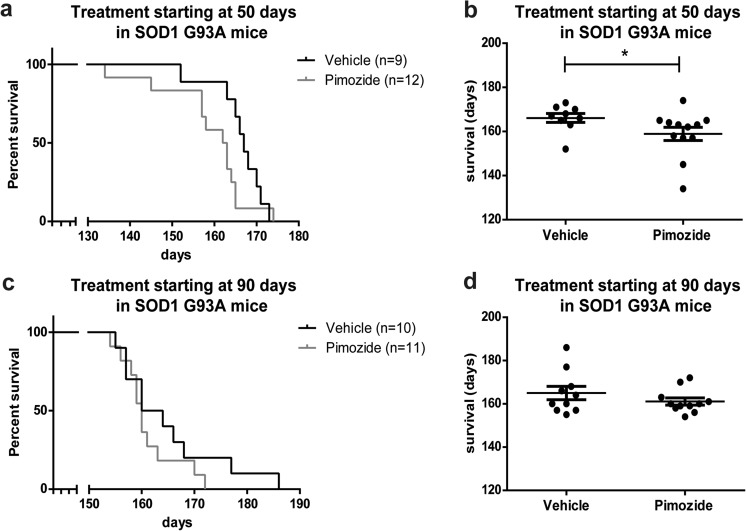
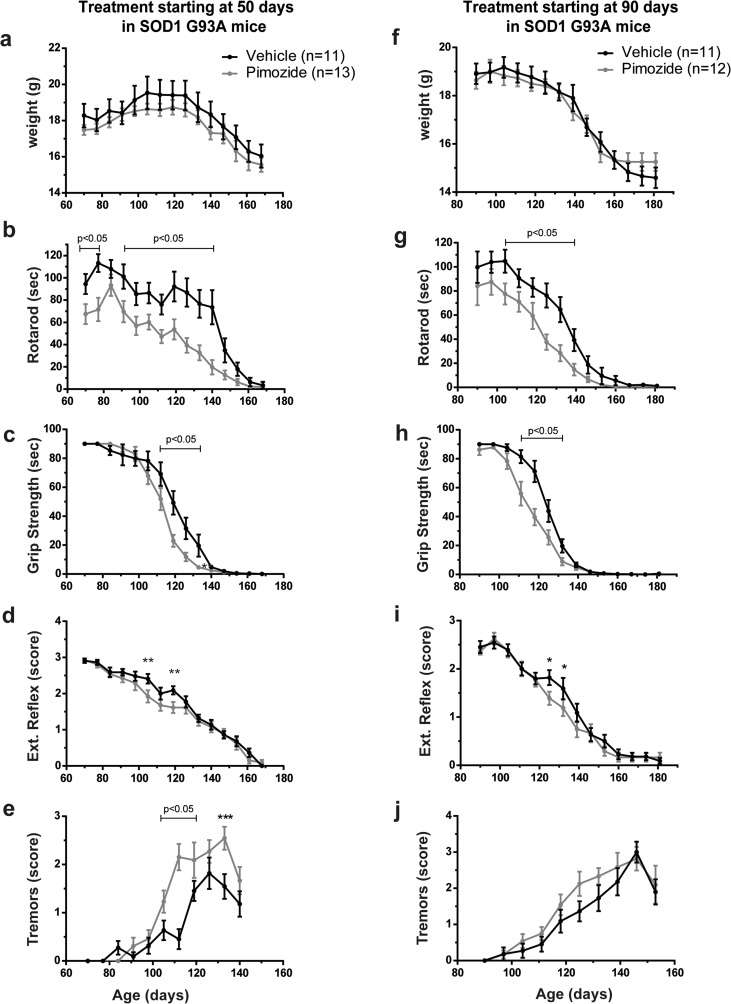
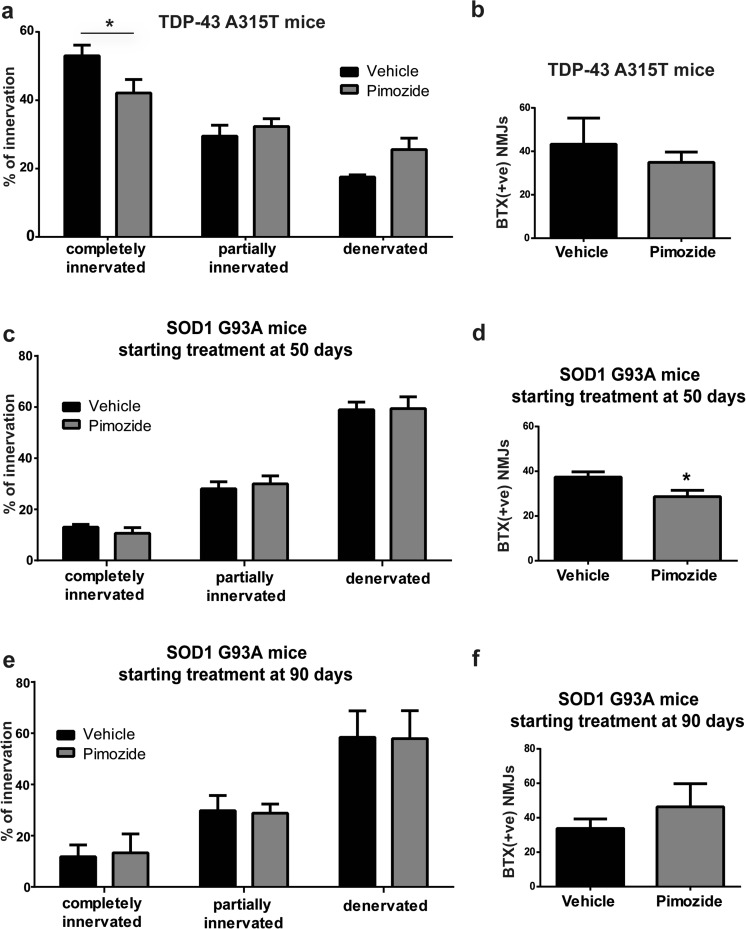
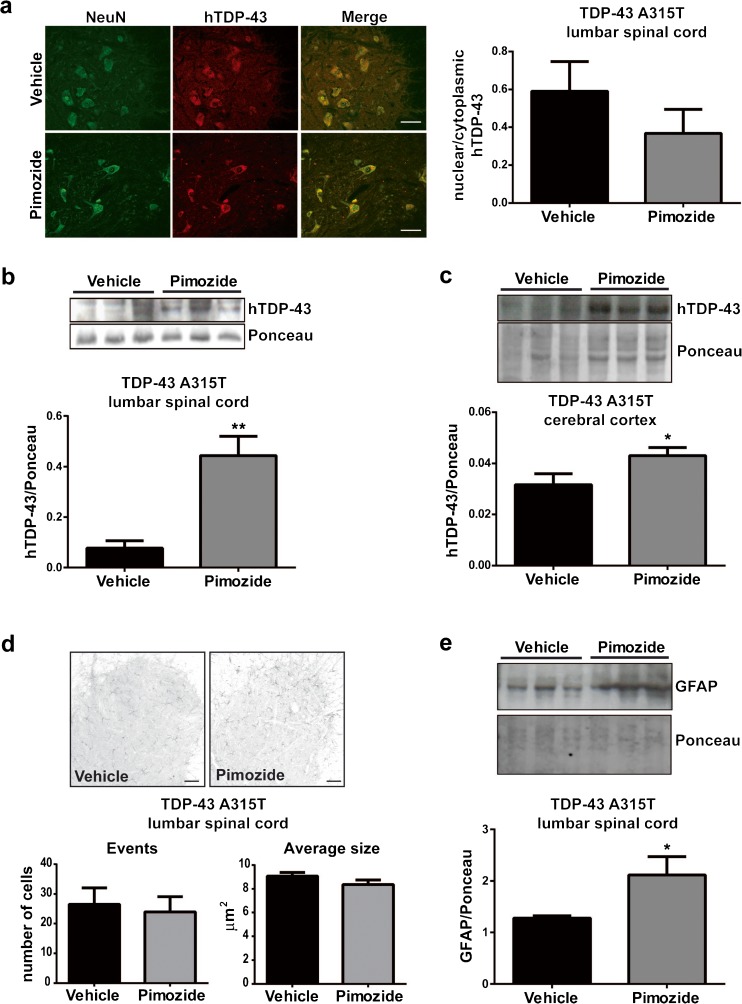
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease which presently does not have any efficient therapeutic approach. Pimozide, a Food and Drug Administration (FDA)-approved neuroepileptic drug, has been recently proposed as a promising treatment for ALS patients based on apparent stabilization of right hand muscles after a short-time administration. A new clinical trial started at the end of 2017 to recruit patients with a prolonged drug delivery schedule. Here, our aim was to investigate the effects of chronic administration of pimozide on disease progression and pathological events in two mouse models of ALS. Pimozide was administered every 2 days to transgenic mice bearing the ALS-linked A315T mutation on the human TAR DNA-binding protein 43 (TDP-43) gene and to mice carrying the human superoxide dismutase 1 (SOD1) gene with the ALS-linked G93A mutation. Chronic administration of pimozide exacerbated motor performances in both animal models and reduced survival in SOD1 mice. In TDP-43T, it decreased the percentage of innervated neuromuscular junctions (NMJs) and increased the accumulation of insoluble TDP-43. In SOD1 mice, pimozide had no effects on NMJ innervation or motoneuron loss, but it increased the levels of misfolded SOD1. We conclude that a chronic administration of pimozide did not confer beneficial effects on disease progression in two mouse models of ALS. In light of a new clinical trial on ALS patients with a chronic regime of pimozide, these results with mouse models suggest prudence and careful monitoring of ALS patients subjected to pimozide treatment.
肌萎缩侧索硬化症(ALS)是一种致命的神经退行性疾病,目前尚无有效的治疗方法。匹莫齐特是一种获得美国食品和药物管理局(FDA)批准的神经抗癫痫药物,最近有研究提出,它可能是 ALS 患者的一种有前途的治疗方法,因为它能在短时间给药后明显稳定右手肌肉。一项新的临床试验于 2017 年底开始招募采用延长药物输送方案的患者。在这里,我们的目的是研究慢性给予匹莫齐特对两种 ALS 小鼠模型疾病进展和病理事件的影响。匹莫齐特每 2 天给药一次,用于携带 ALS 相关 A315T 突变的人类 TAR DNA 结合蛋白 43(TDP-43)基因转基因小鼠和携带 ALS 相关 G93A 突变的人类超氧化物歧化酶 1(SOD1)基因的小鼠。慢性给予匹莫齐特可加重两种动物模型的运动表现,并降低 SOD1 小鼠的存活率。在 TDP-43T 中,它降低了神经肌肉接头(NMJ)的神经支配百分比,并增加了不溶性 TDP-43 的积累。在 SOD1 小鼠中,匹莫齐特对 NMJ 神经支配或运动神经元丢失没有影响,但增加了错误折叠的 SOD1 水平。我们得出的结论是,慢性给予匹莫齐特在两种 ALS 小鼠模型中对疾病进展没有带来有益的影响。鉴于一项关于 ALS 患者的新临床试验,采用慢性匹莫齐特方案,这些用小鼠模型得出的结果表明,对于接受匹莫齐特治疗的 ALS 患者,应该谨慎并仔细监测。