Department of Zoology, University of Oxford, Oxford, UK.
Department of Laboratory Medicine, University of California, San Francisco, San Francisco, CA, USA; UCSF-Abbott Viral Diagnostics and Discovery Center, San Francisco, CA, USA.
Cell Host Microbe. 2018 Jun 13;23(6):855-864.e7. doi: 10.1016/j.chom.2018.04.017. Epub 2018 May 24.
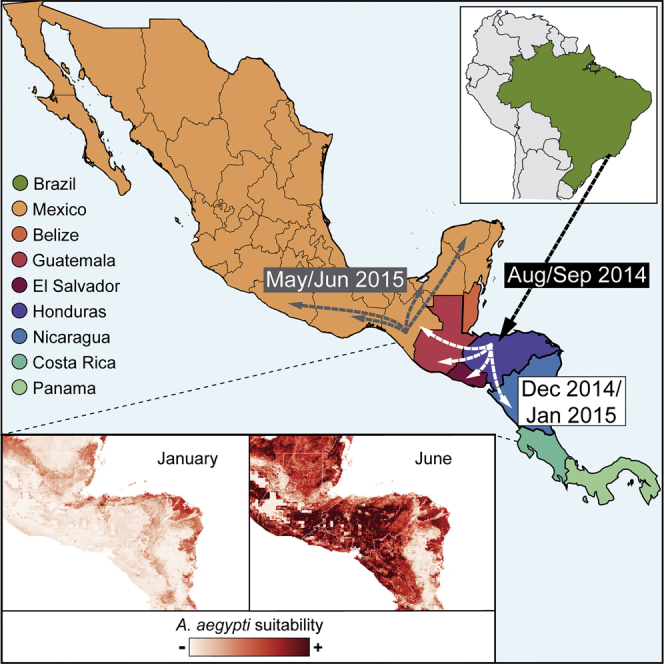
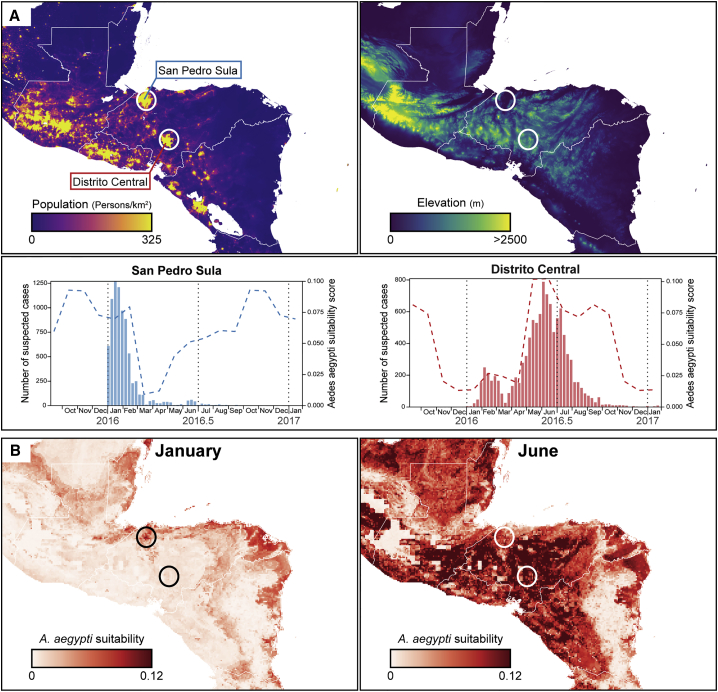
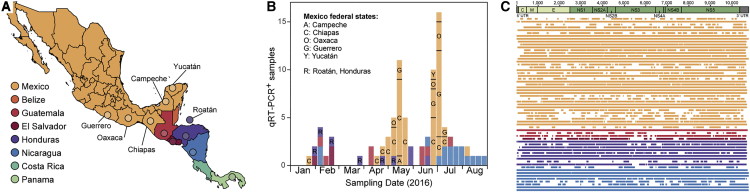
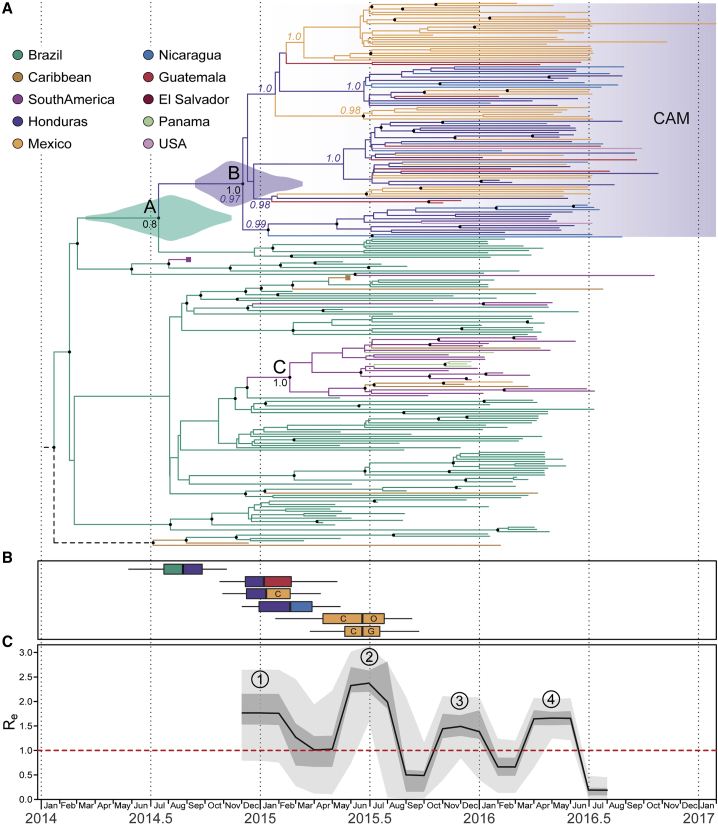
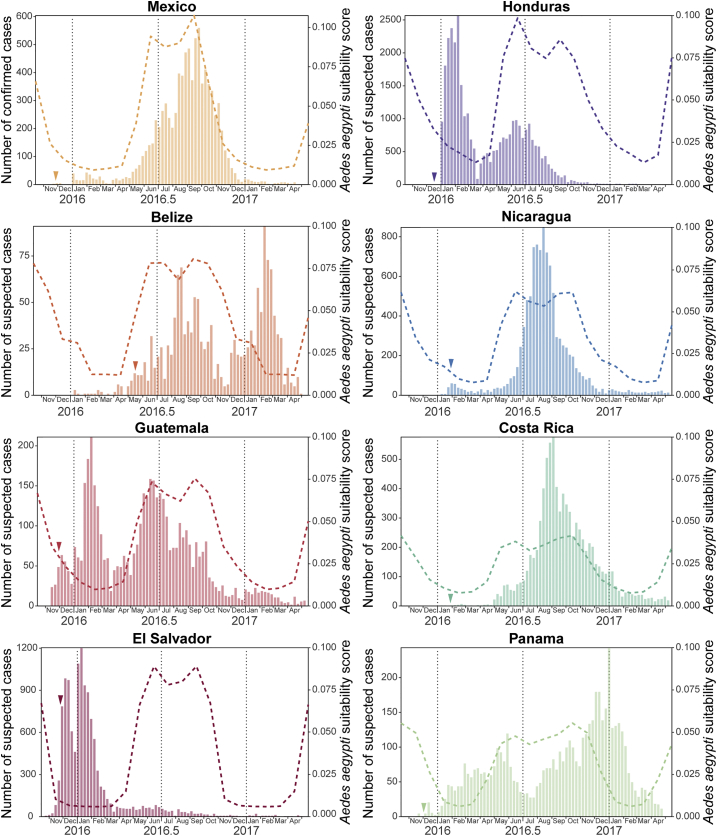
The Zika virus (ZIKV) epidemic in the Americas established ZIKV as a major public health threat and uncovered its association with severe diseases, including microcephaly. However, genetic epidemiology in some at-risk regions, particularly Central America and Mexico, remains limited. We report 61 ZIKV genomes from this region, generated using metagenomic sequencing with ZIKV-specific enrichment, and combine phylogenetic, epidemiological, and environmental data to reconstruct ZIKV transmission. These analyses revealed multiple independent ZIKV introductions to Central America and Mexico. One introduction, likely from Brazil via Honduras, led to most infections and the undetected spread of ZIKV through the region from late 2014. Multiple lines of evidence indicate biannual peaks of ZIKV transmission in the region, likely driven by varying local environmental conditions for mosquito vectors and herd immunity. The spatial and temporal heterogeneity of ZIKV transmission in Central America and Mexico challenges arbovirus surveillance and disease control measures.
Zika 病毒(ZIKV)在美洲的流行确立了 ZIKV 作为一个主要的公共卫生威胁,并揭示了其与包括小头畸形在内的严重疾病的关联。然而,一些高危地区,特别是中美洲和墨西哥的遗传流行病学仍然有限。我们报告了来自该地区的 61 个 ZIKV 基因组,这些基因组是使用具有 ZIKV 特异性富集的宏基因组测序生成的,并结合了系统发生、流行病学和环境数据来重建 ZIKV 的传播。这些分析显示了 ZIKV 多次独立传入中美洲和墨西哥。一次传入,可能是通过洪都拉斯从巴西传入,导致了大部分感染,并在 2014 年底之前在该地区传播了 ZIKV。多种证据表明,该地区 ZIKV 传播存在双年高峰,可能是由蚊子媒介和群体免疫的当地环境条件的变化驱动的。中美洲和墨西哥 ZIKV 传播的空间和时间异质性对虫媒病毒监测和疾病控制措施提出了挑战。