European Research Institute for the Biology of Ageing, University of Groningen, University Medical Centre Groningen, A. Deusinglaan 1, Groningen, NL-9713 AV, The Netherlands.
Institute of Computational Biology, Helmholtz Zentrum München, Ingolstädter Landstr. 1, Neuherberg, 85764, Germany.
BMC Genomics. 2018 Jun 7;19(1):444. doi: 10.1186/s12864-018-4641-x.
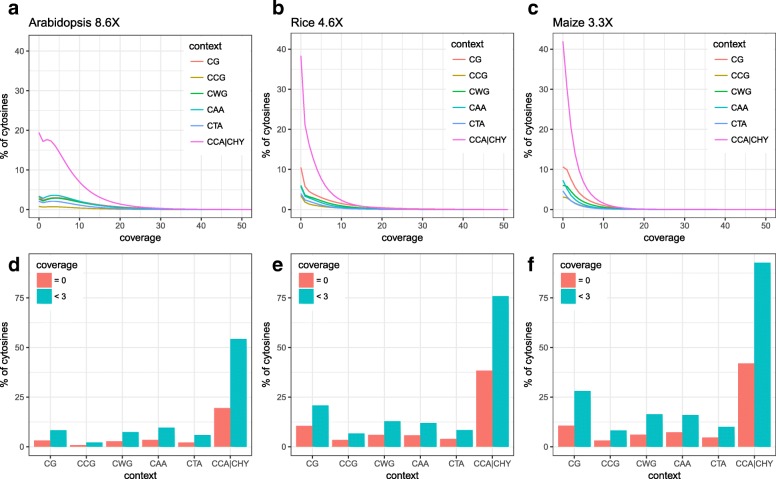
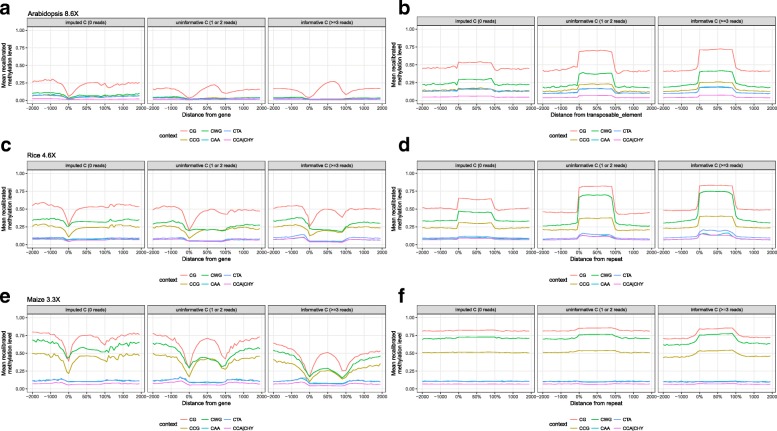
Whole-genome bisulfite sequencing (WGBS) has become the standard method for interrogating plant methylomes at base resolution. However, deep WGBS measurements remain cost prohibitive for large, complex genomes and for population-level studies. As a result, most published plant methylomes are sequenced far below saturation, with a large proportion of cytosines having either missing data or insufficient coverage.
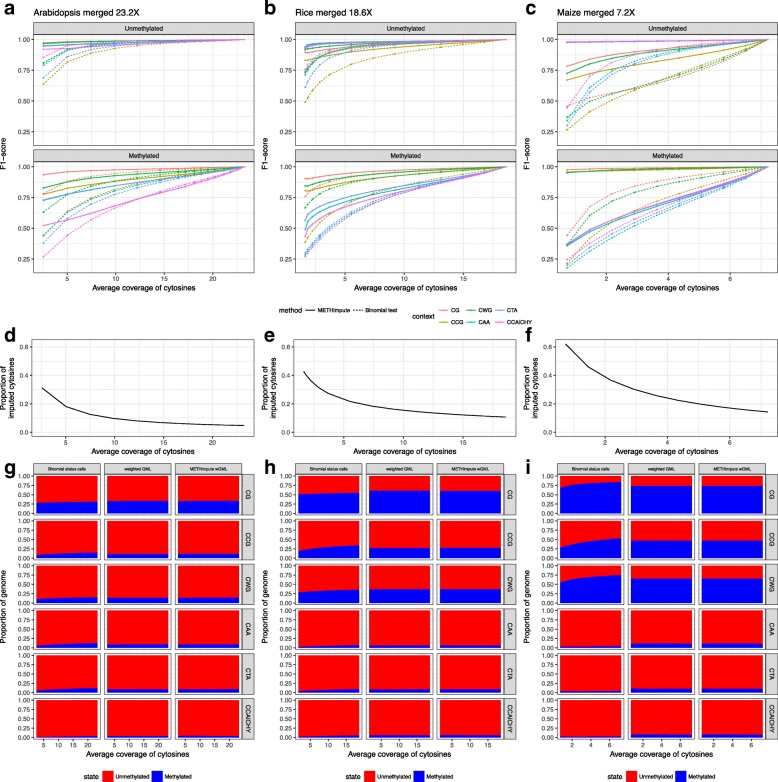
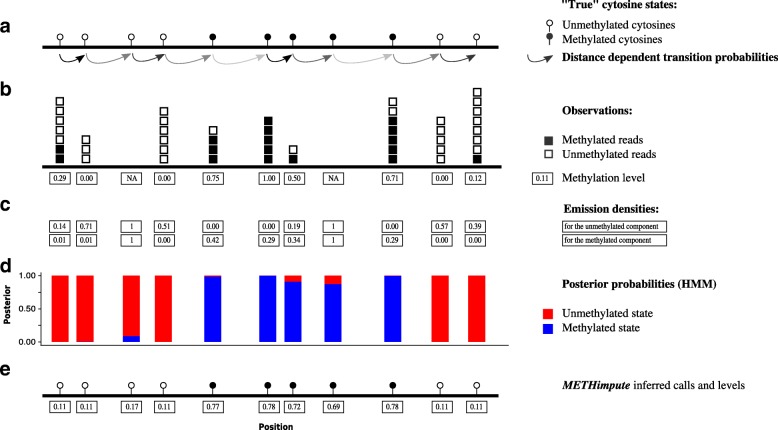
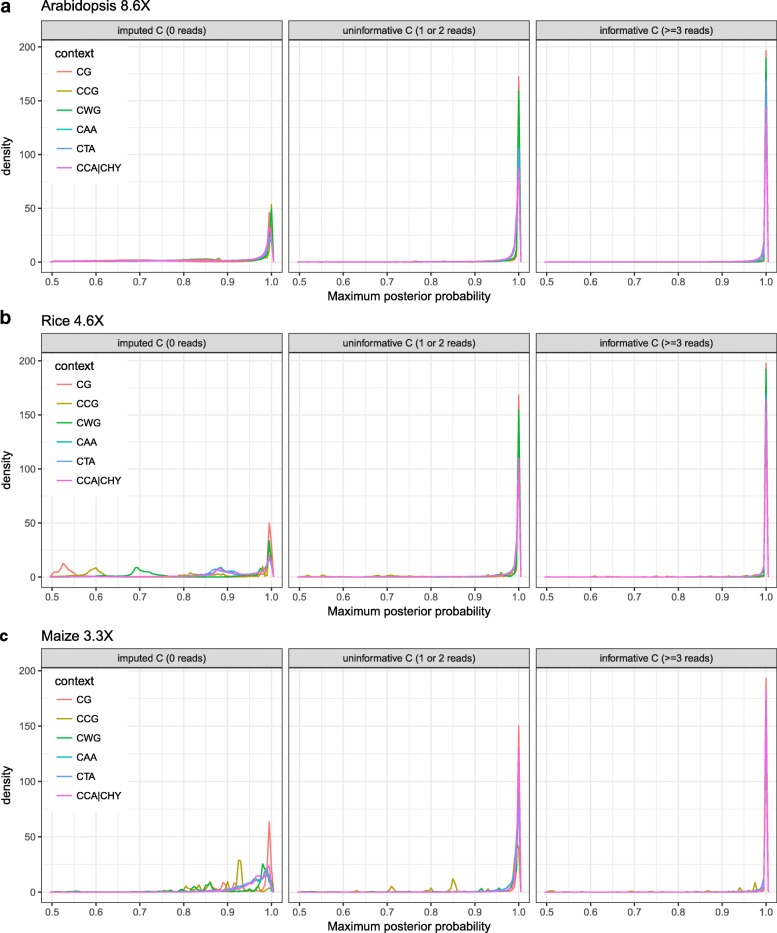
Here we present METHimpute, a Hidden Markov Model (HMM) based imputation algorithm for the analysis of WGBS data. Unlike existing methods, METHimpute enables the construction of complete methylomes by inferring the methylation status and level of all cytosines in the genome regardless of coverage. Application of METHimpute to maize, rice and Arabidopsis shows that the algorithm infers cytosine-resolution methylomes with high accuracy from data as low as 6X, compared to data with 60X, thus making it a cost-effective solution for large-scale studies.
METHimpute provides methylation status calls and levels for all cytosines in the genome regardless of coverage, thus yielding complete methylomes even with low-coverage WGBS datasets. The method has been extensively tested in plants, but should also be applicable to other species. An implementation is available on Bioconductor.
全基因组亚硫酸氢盐测序(WGBS)已成为解析植物甲基组的标准方法。然而,对于大型复杂基因组和群体水平的研究来说,深度 WGBS 测量仍然成本过高。因此,大多数已发表的植物甲基组学研究都是在远远低于饱和的深度下进行测序的,大量的胞嘧啶要么数据缺失,要么覆盖度不足。
这里我们提出了 METHimpute,这是一种基于隐马尔可夫模型(HMM)的 WGBS 数据分析的插补算法。与现有方法不同,METHimpute 能够通过推断基因组中所有胞嘧啶的甲基化状态和水平来构建完整的甲基组,而不管覆盖度如何。将 METHimpute 应用于玉米、水稻和拟南芥表明,与 60X 数据相比,该算法能够从低至 6X 的数据中以高精度推断出具有胞嘧啶分辨率的甲基组,因此对于大规模研究来说是一种具有成本效益的解决方案。
METHimpute 提供了基因组中所有胞嘧啶的甲基化状态和水平的调用,因此即使在低覆盖度的 WGBS 数据集上也能生成完整的甲基组。该方法已在植物中进行了广泛的测试,但也应该适用于其他物种。该方法的实现可在 Bioconductor 上获得。