IBM T. J. Watson Research Center, Yorktown Heights, New York, United States of America.
Physics Institute - University of Brasilia, Brasilia, Brazil.
PLoS Comput Biol. 2018 Jul 2;14(7):e1006305. doi: 10.1371/journal.pcbi.1006305. eCollection 2018 Jul.
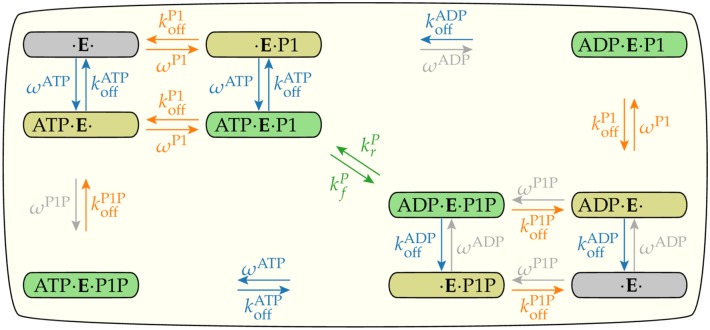
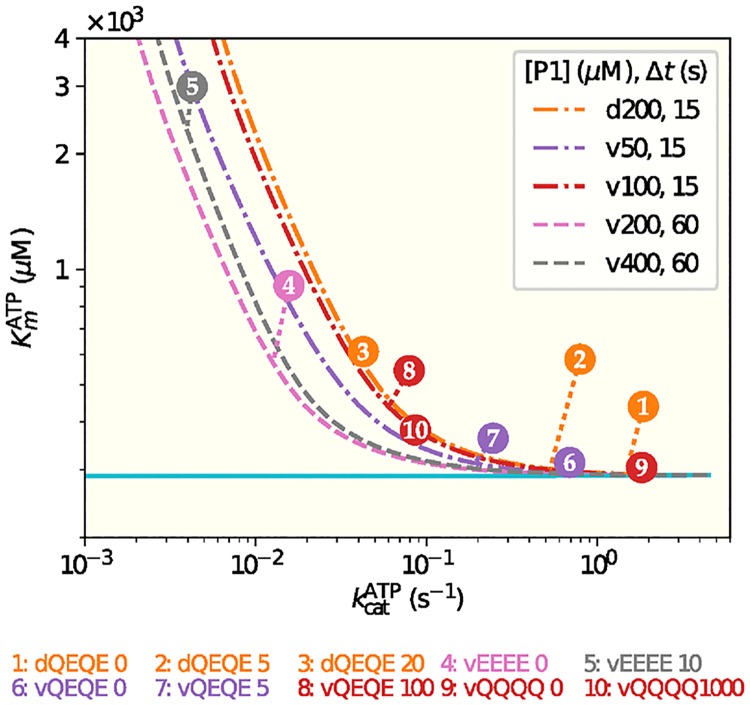
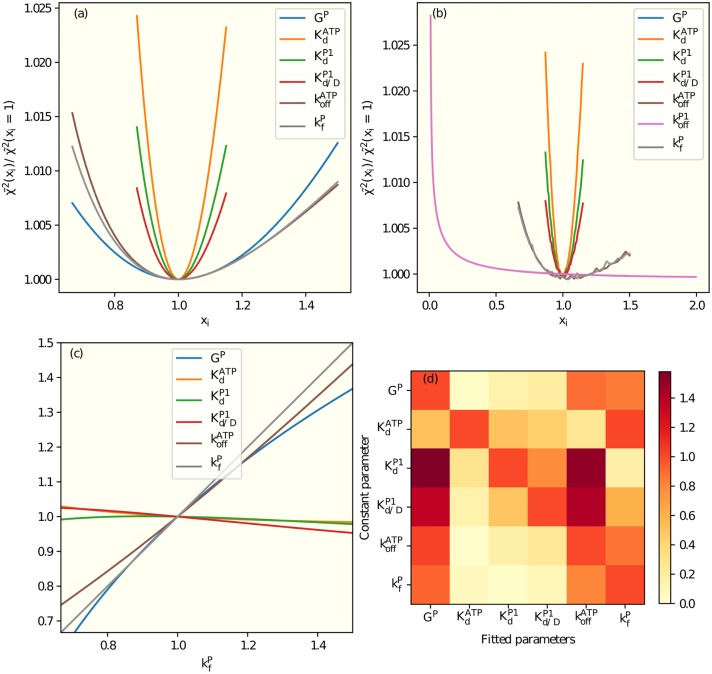
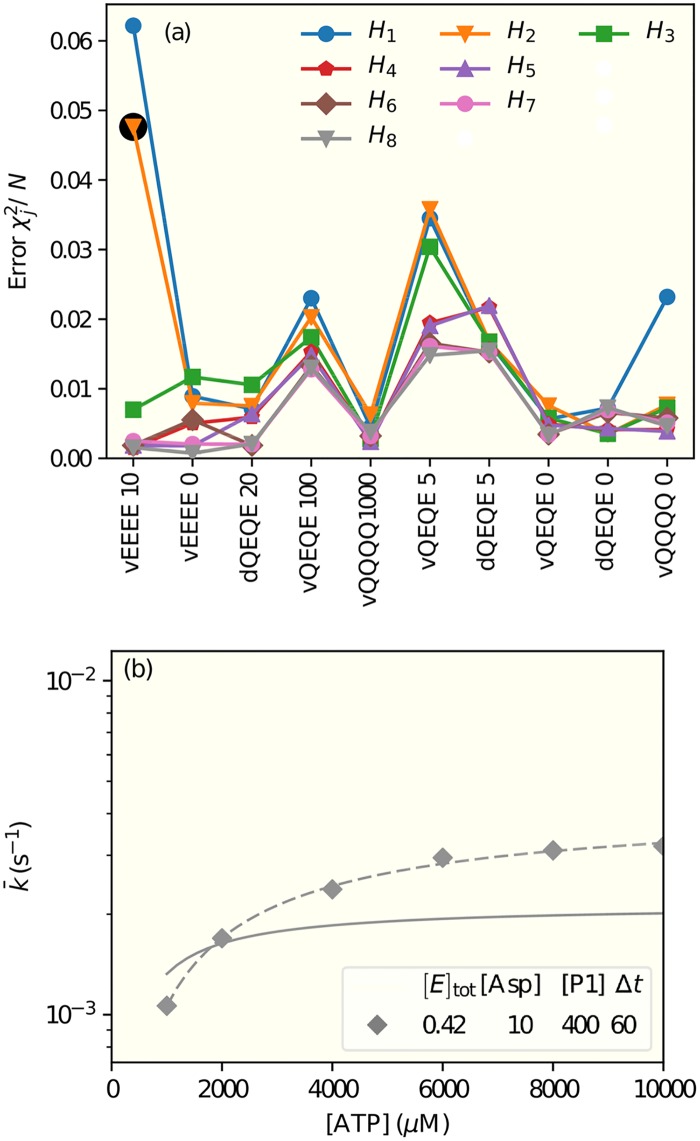
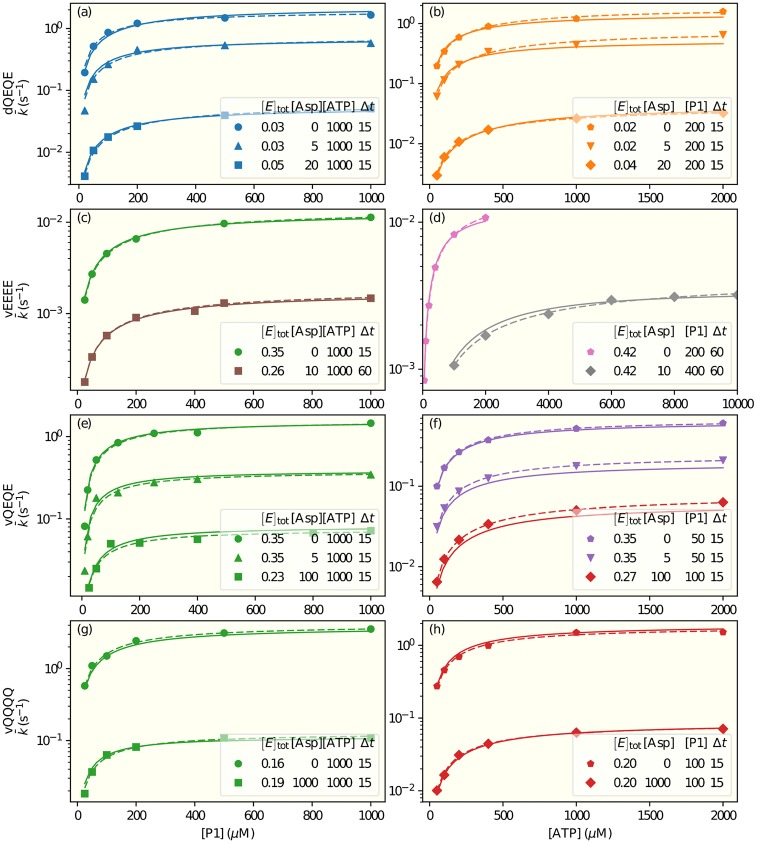
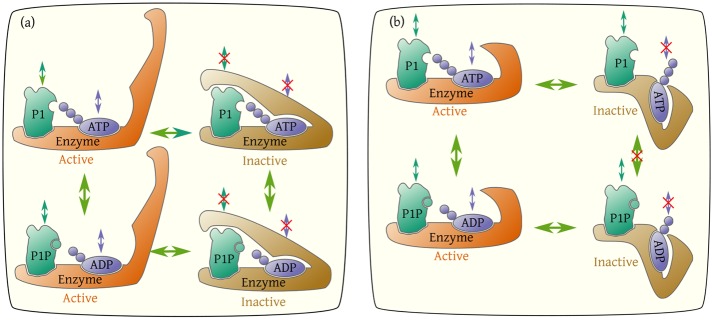
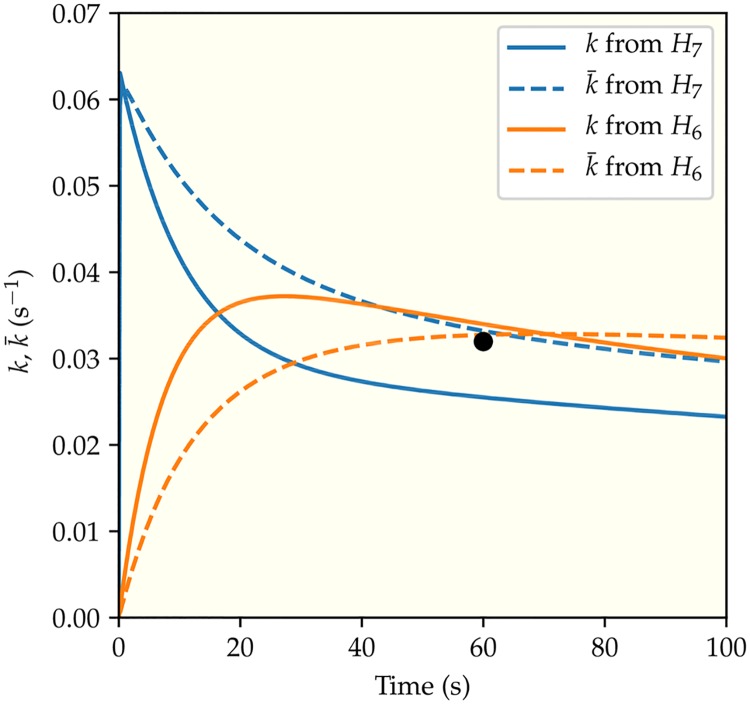
It is challenging to decipher molecular mechanisms in biological systems from system-level input-output data, especially for complex processes that involve interactions among multiple components. We addressed this general problem for the bacterial histidine kinase CheA, the activity of which is regulated in chemotaxis signaling complexes by bacterial chemoreceptors. We developed a general network model to describe the dynamics of the system, treating the receptor complex with coupling protein CheW and the P3P4P5 domains of kinase CheA as a regulated enzyme with two substrates, ATP and P1, the phosphoryl-accepting domain of CheA. Our simple network model allowed us to search hypothesis space systematically. For different and progressively more complex regulation schemes, we fit our models to a large set of input-output data with the aim of identifying the simplest possible regulation mechanisms consistent with the data. Our modeling and analysis revealed novel dual regulation mechanisms in which receptor activity regulated ATP binding plus one other process, either P1 binding or phosphoryl transfer between P1 and ATP. Strikingly, in our models receptor control affected the kinetic rate constants of substrate association and dissociation equally and thus did not alter the respective equilibrium constants. We suggest experiments that could distinguish between the two dual-regulation mechanisms. This systems-biology approach of combining modeling and a large input-output dataset should be applicable for studying other complex biological processes.
从系统层面的输入-输出数据中破译生物系统的分子机制极具挑战性,特别是对于涉及多个组件相互作用的复杂过程而言。我们针对细菌组氨酸激酶 CheA 的这一普遍问题进行了研究,该激酶的活性在趋化信号复合物中受到细菌化学感受器的调节。我们开发了一种通用的网络模型来描述系统的动态,将受体复合物与偶联蛋白 CheW 和激酶 CheA 的 P3P4P5 结构域视为一种具有两个底物(ATP 和 P1)的调节酶,其中 P1 是 CheA 的磷酸受体结构域。我们的简单网络模型使我们能够系统地搜索假说空间。对于不同且越来越复杂的调节方案,我们将模型拟合到大量输入-输出数据中,目的是确定与数据一致的最简单可能的调节机制。我们的建模和分析揭示了新的双重调节机制,其中受体活性调节 ATP 结合以及另一个过程,要么是 P1 结合,要么是 P1 和 ATP 之间的磷酸转移。引人注目的是,在我们的模型中,受体控制对底物结合和解离的动力学速率常数的影响相同,因此不会改变各自的平衡常数。我们建议进行一些实验,以区分这两种双重调节机制。这种结合建模和大量输入-输出数据集的系统生物学方法应该适用于研究其他复杂的生物过程。