Ajani Haresh, Pecina Adam, Eyrilmez Saltuk M, Fanfrlík Jindřich, Haldar Susanta, Řezáč Jan, Hobza Pavel, Lepšík Martin
Department of Computational Chemistry, Institute of Organic Chemistry and Biochemistry, Czech Academy of Sciences, v.v.i., Flemingovo nam. 2, 16610 Praha 6, Czech Republic.
Department of Physical Chemistry, Palacký University, tř. 17. listopadu 1192/12, 77146 Olomouc, Czech Republic.
ACS Omega. 2017 Jul 31;2(7):4022-4029. doi: 10.1021/acsomega.7b00503. Epub 2017 Jul 27.
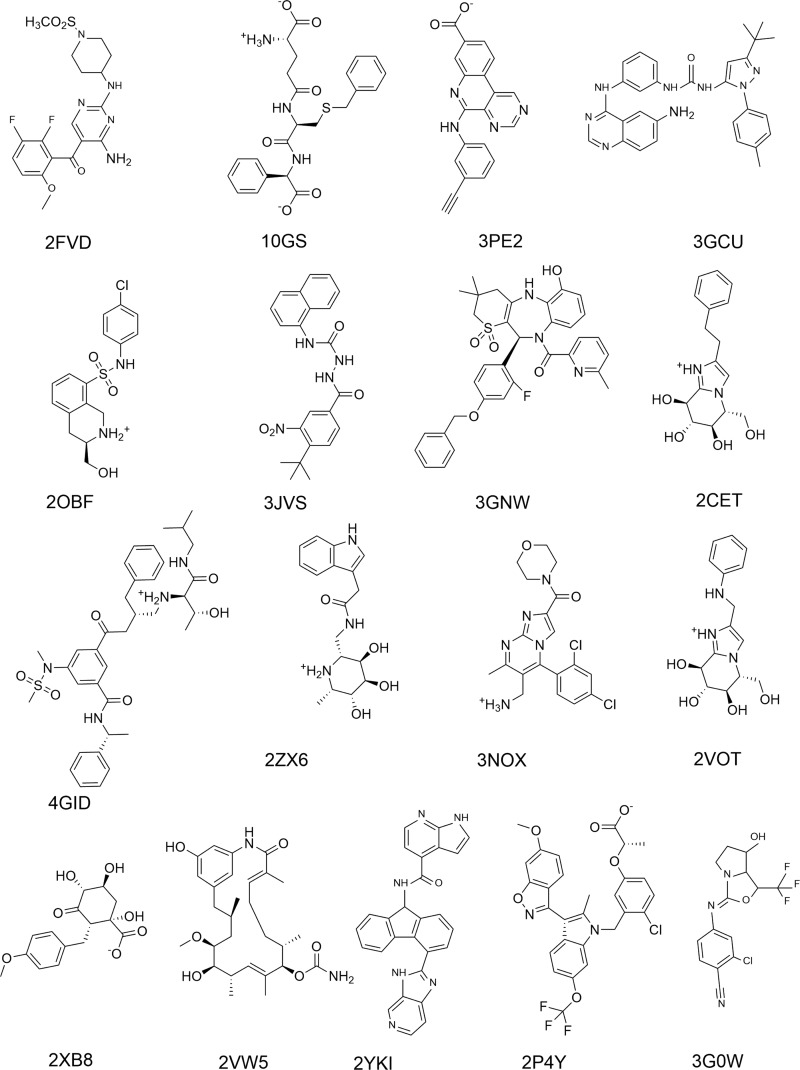
General and reliable description of structures and energetics in protein-ligand (PL) binding using the docking/scoring methodology has until now been elusive. We address this urgent deficiency of scoring functions (SFs) by the systematic development of corrected semiempirical quantum mechanical (SQM) methods, which correctly describe all types of noncovalent interactions and are fast enough to treat systems of thousands of atoms. Two most accurate SQM methods, PM6-D3H4X and SCC-DFTB3-D3H4X, are coupled with the conductor-like screening model (COSMO) implicit solvation model in so-called "SQM/COSMO" SFs and have shown unique recognition of native ligand poses in cognate docking in four challenging PL systems, including metalloprotein. Here, we apply the two SQM/COSMO SFs to 17 diverse PL complexes and compare their performance with four widely used classical SFs (Glide XP, AutoDock4, AutoDock Vina, and UCSF Dock). We observe superior performance of the SQM/COSMO SFs and identify challenging systems. This method, due to its generality, comparability across the chemical space, and lack of need for any system-specific parameters, gives promise of becoming, after comprehensive large-scale testing in the near future, a useful computational tool in structure-based drug design and serving as a reference method for the development of other SFs.
迄今为止,使用对接/评分方法对蛋白质-配体(PL)结合中的结构和能量进行全面且可靠的描述一直难以实现。我们通过系统开发校正的半经验量子力学(SQM)方法来解决评分函数(SFs)这一紧迫的缺陷,该方法能够正确描述所有类型的非共价相互作用,并且速度足够快,可以处理数千个原子的系统。两种最精确的SQM方法,即PM6-D3H4X和SCC-DFTB3-D3H4X,与类导体屏蔽模型(COSMO)隐式溶剂化模型相结合,形成所谓的“SQM/COSMO”评分函数,并已在包括金属蛋白在内的四个具有挑战性的PL系统的同源对接中显示出对天然配体构象的独特识别能力。在此,我们将这两种SQM/COSMO评分函数应用于17种不同的PL复合物,并将它们的性能与四种广泛使用的经典评分函数(Glide XP、AutoDock4、AutoDock Vina和UCSF Dock)进行比较。我们观察到SQM/COSMO评分函数具有卓越的性能,并识别出具有挑战性的系统。由于该方法具有通用性、在化学空间中的可比性以及无需任何特定于系统的参数,有望在不久的将来经过全面的大规模测试后,成为基于结构的药物设计中一种有用的计算工具,并作为开发其他评分函数的参考方法。