Gupta Sanjana, Hainsworth Liam, Hogg Justin S, Lee Robin E C, Faeder James R
Department of Computational and Systems Biology, University of Pittsburgh School of Medicine, Pittsburgh, PA 15260, USA.
Proc Euromicro Int Conf Parallel Distrib Netw Based Process. 2018 Mar;2018:690-697. doi: 10.1109/PDP2018.2018.00114. Epub 2018 Jun 7.
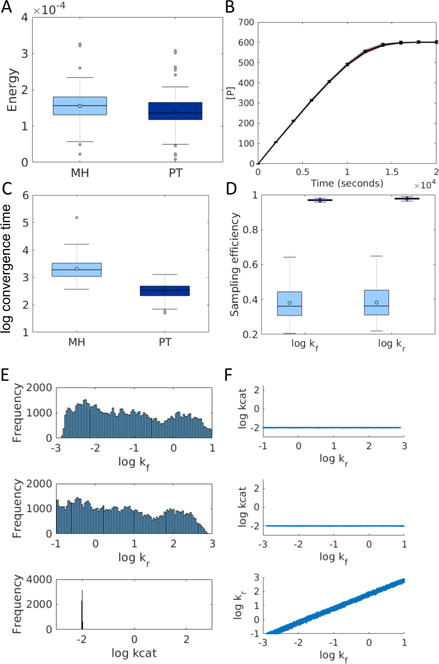
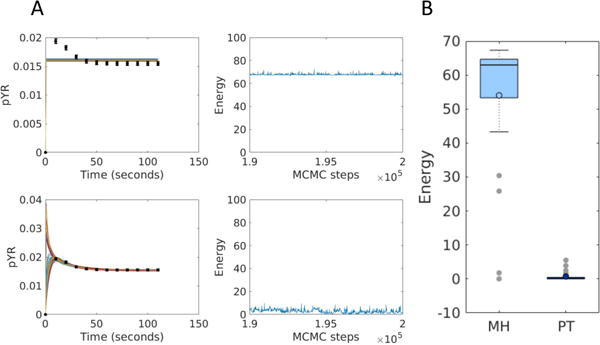
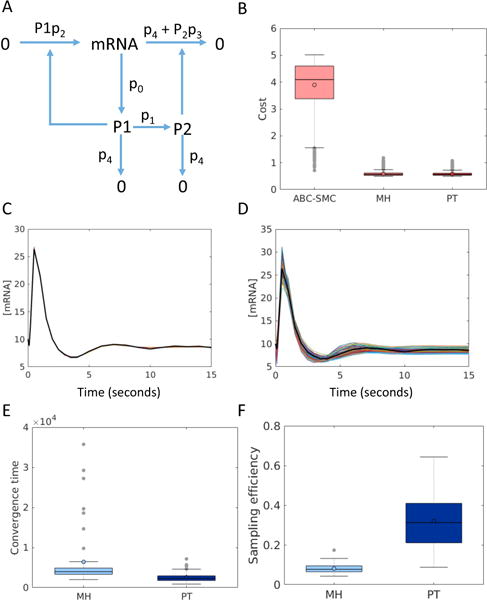
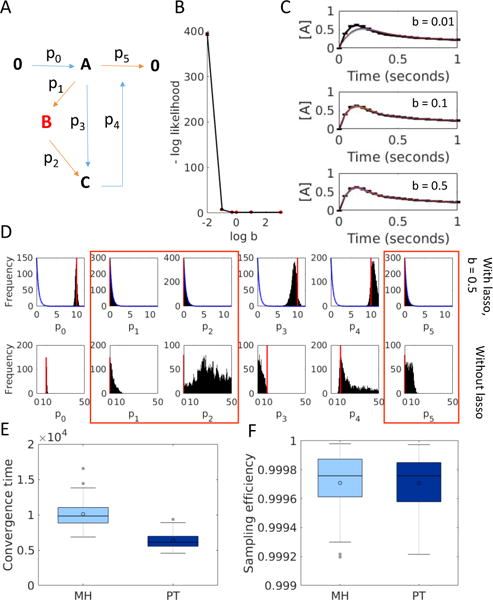
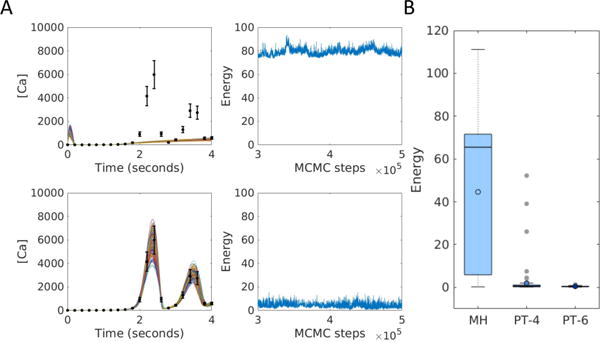
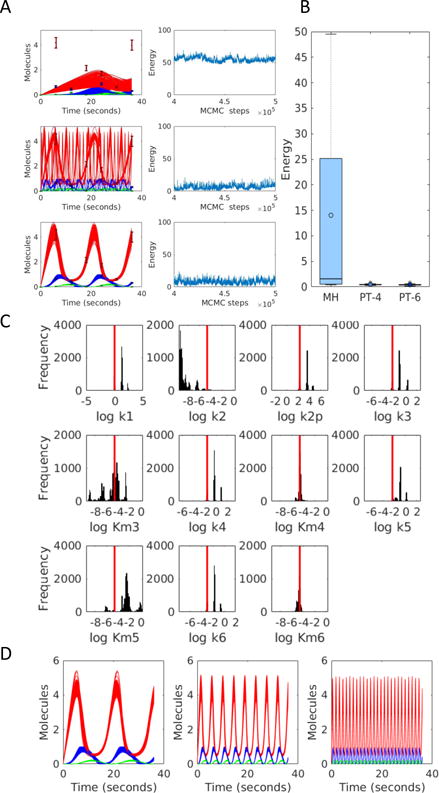
Models of biological systems often have many unknown parameters that must be determined in order for model behavior to match experimental observations. Commonly-used methods for parameter estimation that return point estimates of the best-fit parameters are insufficient when models are high dimensional and under-constrained. As a result, Bayesian methods, which treat model parameters as random variables and attempt to estimate their probability distributions given data, have become popular in systems biology. Bayesian parameter estimation often relies on Markov Chain Monte Carlo (MCMC) methods to sample model parameter distributions, but the slow convergence of MCMC sampling can be a major bottleneck. One approach to improving performance is parallel tempering (PT), a physics-based method that uses swapping between multiple Markov chains run in parallel at different temperatures to accelerate sampling. The temperature of a Markov chain determines the probability of accepting an unfavorable move, so swapping with higher temperatures chains enables the sampling chain to escape from local minima. In this work we compared the MCMC performance of PT and the commonly-used Metropolis-Hastings (MH) algorithm on six biological models of varying complexity. We found that for simpler models PT accelerated convergence and sampling, and that for more complex models, PT often converged in cases MH became trapped in non-optimal local minima. We also developed a freely-available MATLAB package for Bayesian parameter estimation called PTEMPEST (http://github.com/RuleWorld/ptempest), which is closely integrated with the popular BioNetGen software for rule-based modeling of biological systems.
生物系统模型通常有许多未知参数,为使模型行为与实验观测结果相匹配,必须确定这些参数。当模型维度高且约束不足时,常用的返回最佳拟合参数点估计的参数估计方法并不充分。因此,将模型参数视为随机变量并尝试根据数据估计其概率分布的贝叶斯方法在系统生物学中变得很流行。贝叶斯参数估计通常依赖马尔可夫链蒙特卡罗(MCMC)方法对模型参数分布进行采样,但MCMC采样的缓慢收敛可能是一个主要瓶颈。一种提高性能的方法是并行回火(PT),这是一种基于物理的方法,它使用在不同温度下并行运行的多个马尔可夫链之间的交换来加速采样。马尔可夫链的温度决定了接受不利移动的概率,因此与较高温度的链进行交换可使采样链逃离局部最小值。在这项工作中,我们比较了PT和常用的梅特罗波利斯-黑斯廷斯(MH)算法在六个不同复杂程度的生物模型上的MCMC性能。我们发现,对于较简单的模型,PT加速了收敛和采样,而对于更复杂的模型,PT在很多情况下能收敛,而MH则陷入非最优局部最小值。我们还开发了一个名为PTEMPEST(http://github.com/RuleWorld/ptempest)的用于贝叶斯参数估计的免费MATLAB软件包,它与用于生物系统基于规则建模的流行软件BioNetGen紧密集成。