Shi Ying, Kellingray Lee, Zhai Qixiao, Gall Gwenaelle Le, Narbad Arjan, Zhao Jianxin, Zhang Hao, Chen Wei
State Key Laboratory of Food Science and Technology, Jiangnan University, Wuxi, China.
National Engineering Research Center for Functional Food, Wuxi, China.
Front Microbiol. 2018 Aug 21;9:1948. doi: 10.3389/fmicb.2018.01948. eCollection 2018.
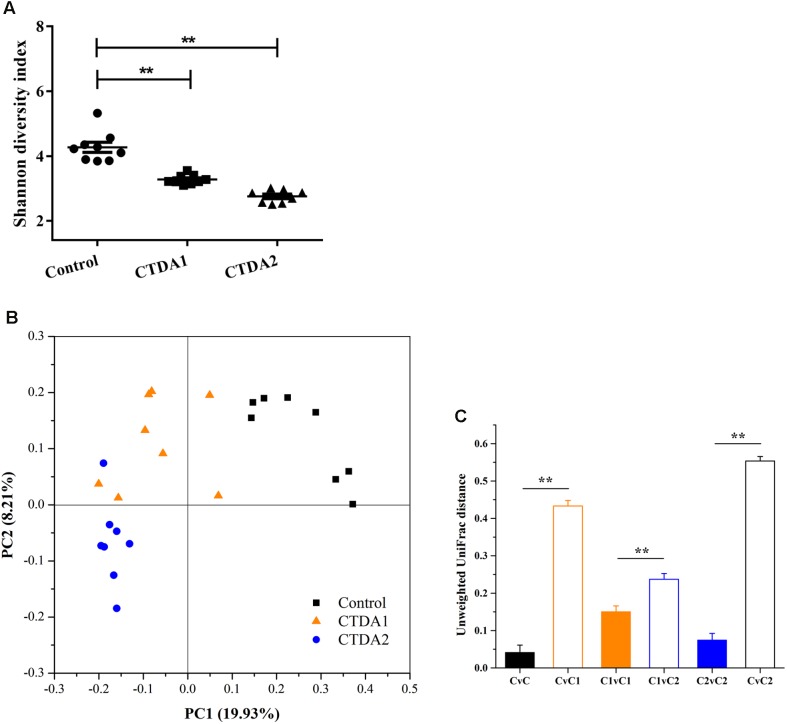
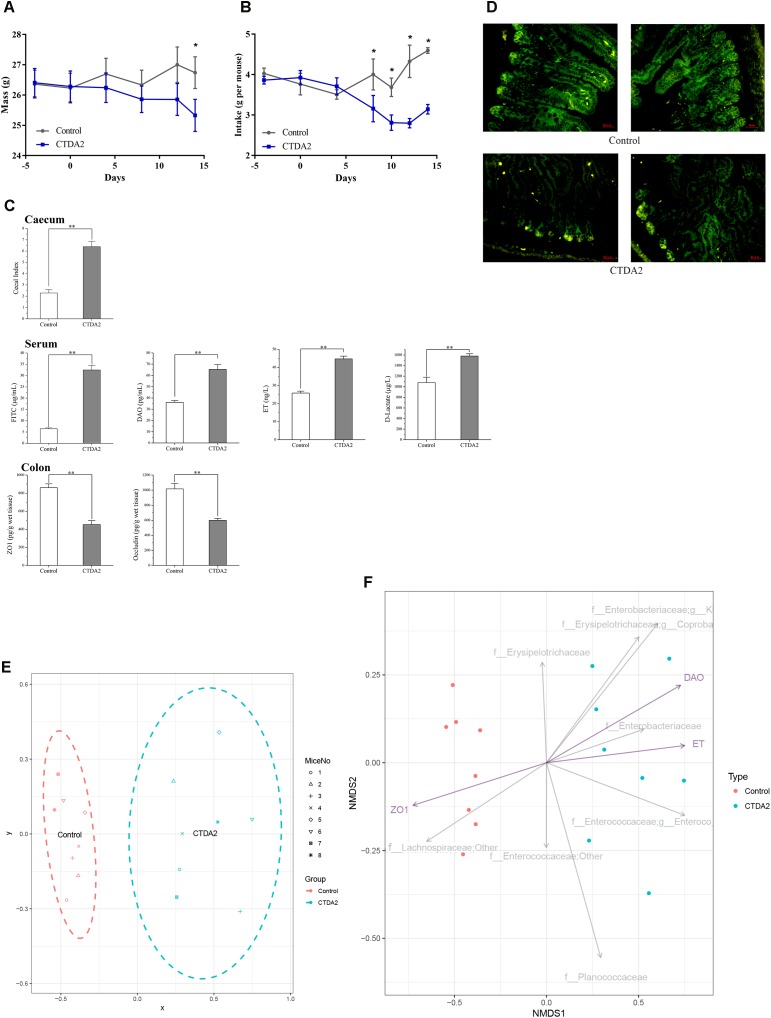
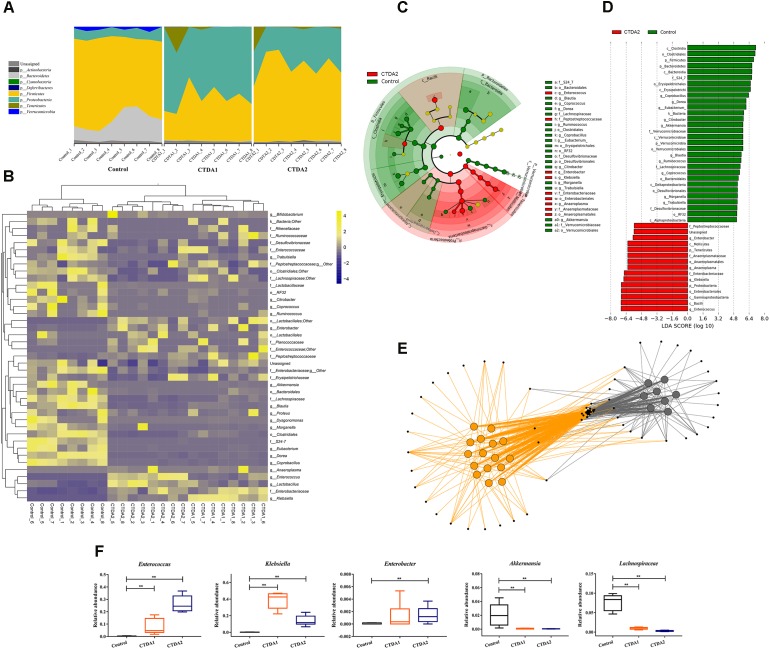
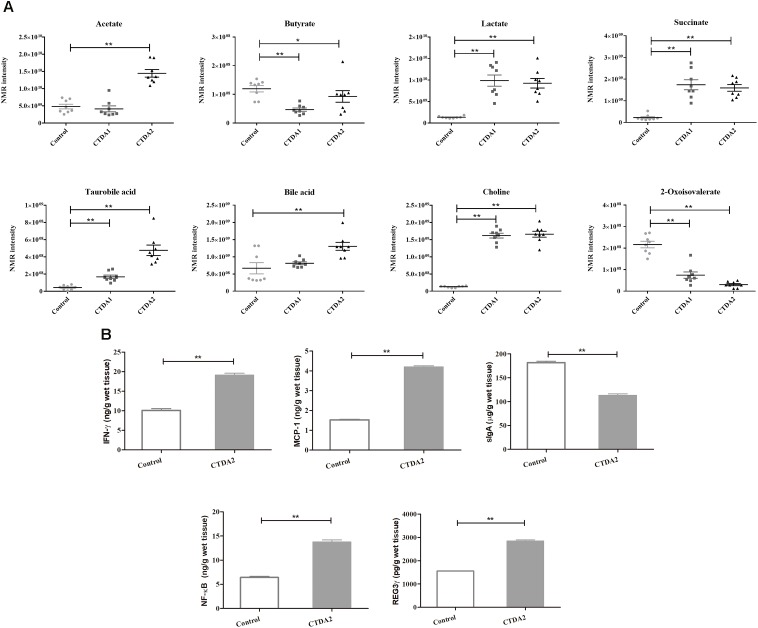
The aim of this study was to establish continuous therapeutic-dose ampicillin (CTDA)-induced dysbiosis in a mouse model, mimicking typical adult exposure, with a view to using this to assess its impact on gut microbiota, intestinal metabolites and host immune responses. Mice were exposed to ampicillin for 14 days and antibiotic-induced dysbiosis was evaluated by alteration of microbiota and gut permeability. The cecal index was increased in the CTDA group, and the gut permeability indicated by fluorescent dextran, endotoxin and D-Lactate in the serum was significantly increased after antibiotic use. The tight-junction proteins ZO-1 and occludin in the colon were reduced to half the control level in CTDA. We found that alpha-diversity was significantly decreased in mice receiving CTDA, and microbial community structure was altered compared with the control. Key taxa were identified as CTDA-specific, and the relative abundance of and was particularly enriched while , and were depleted after antibiotic treatment. In particular, a significant increase in succinate and a reduction in butyrate was detected in CTDA mice, and the triggering of NF-κB enhancement reflected that the host immune response was influenced by ampicillin use. The observed perturbation of the microbiota was accompanied by modulation of inflammatory state; this included increase in interferon-γ and RegIIIγ, and a decrease in secretory IgA in the colon mucosa. This study allowed us to identify the key taxa associated with an ampicillin-induced state of dysbiosis in mice and to characterize the microbial communities via molecular profiling. Thus, this work describes the bacterial ecology of antibiotic exposure model in combination with host physiological characteristics at a detailed level of microbial taxa.
本研究的目的是在模仿典型成人暴露情况的小鼠模型中建立连续治疗剂量氨苄西林(CTDA)诱导的菌群失调,以便利用其评估对肠道微生物群、肠道代谢物和宿主免疫反应的影响。将小鼠暴露于氨苄西林14天,并通过微生物群和肠道通透性的改变来评估抗生素诱导的菌群失调。CTDA组的盲肠指数增加,使用抗生素后,血清中荧光葡聚糖、内毒素和D-乳酸所表明的肠道通透性显著增加。CTDA组中结肠的紧密连接蛋白ZO-1和闭合蛋白减少至对照水平的一半。我们发现,接受CTDA的小鼠的α多样性显著降低,与对照组相比,微生物群落结构发生了改变。确定了关键分类群为CTDA特异性分类群,抗生素治疗后, 和 的相对丰度特别富集,而 、 和 则减少。特别是,在CTDA小鼠中检测到琥珀酸盐显著增加,丁酸盐减少,NF-κB增强的触发反映了宿主免疫反应受到氨苄西林使用的影响。观察到的微生物群扰动伴随着炎症状态的调节;这包括结肠黏膜中干扰素-γ和RegIIIγ增加,分泌型IgA减少。这项研究使我们能够识别与小鼠氨苄西林诱导的菌群失调状态相关的关键分类群,并通过分子谱分析对微生物群落进行表征。因此,这项工作在微生物分类群的详细层面上描述了抗生素暴露模型的细菌生态学与宿主生理特征的结合。