Division of Immunology, Department of Pathology, Johns Hopkins University, Baltimore, MD, USA.
Division of Rheumatology, Department of Medicine, Johns Hopkins University, Baltimore, MD, USA.
Nat Protoc. 2018 Sep;13(9):1958-1978. doi: 10.1038/s41596-018-0025-6.
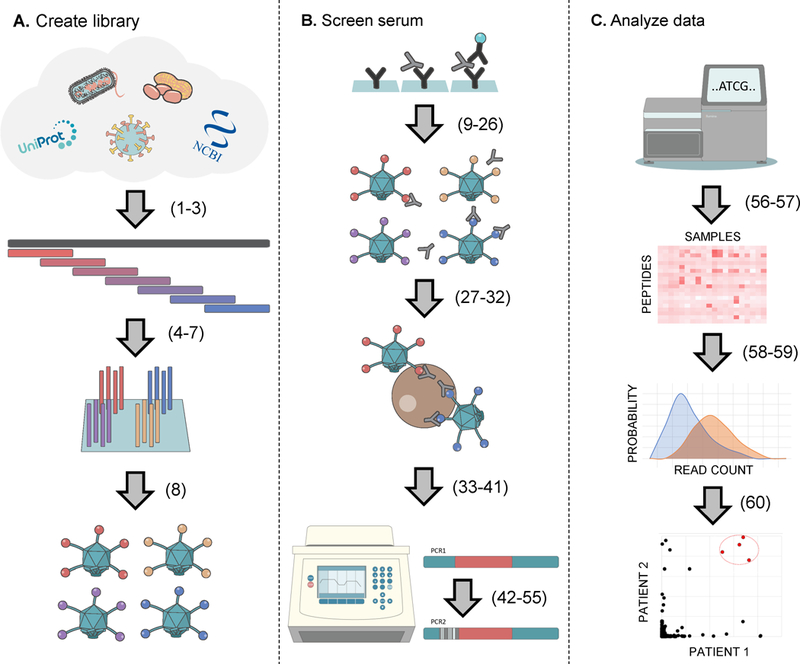
The binding specificities of an individual's antibody repertoire contain a wealth of biological information. They harbor evidence of environmental exposures, allergies, ongoing or emerging autoimmune disease processes, and responses to immunomodulatory therapies, for example. Highly multiplexed methods to comprehensively interrogate antibody-binding specificities have therefore emerged in recent years as important molecular tools. Here, we provide a detailed protocol for performing 'phage immunoprecipitation sequencing' (PhIP-Seq), which is a powerful method for analyzing antibody-repertoire binding specificities with high throughput and at low cost. The methodology uses oligonucleotide library synthesis (OLS) to encode proteomic-scale peptide libraries for display on bacteriophage. These libraries are then immunoprecipitated, using an individual's antibodies, for subsequent analysis by high-throughput DNA sequencing. We have used PhIP-Seq to identify novel self-antigens associated with autoimmune disease, to characterize the self-reactivity of broadly neutralizing HIV antibodies, and in a large international cross-sectional study of exposure to hundreds of human viruses. Compared with alternative array-based techniques, PhIP-Seq is far more scalable in terms of sample throughput and cost per analysis. Cloning and expression of recombinant proteins are not required (versus protein microarrays), and peptide lengths are limited only by DNA synthesis chemistry (up to 90-aa (amino acid) peptides versus the typical 8- to 12-aa length limit of synthetic peptide arrays). Compared with protein microarrays, however, PhIP-Seq libraries lack discontinuous epitopes and post-translational modifications. To increase the accessibility of PhIP-Seq, we provide detailed instructions for the design of phage-displayed peptidome libraries, their immunoprecipitation using serum antibodies, deep sequencing-based measurement of peptide abundances, and statistical determination of peptide enrichments that reflect antibody-peptide interactions. Once a library has been constructed, PhIP-Seq data can be obtained for analysis within a week.
个体抗体库的结合特异性包含丰富的生物学信息。例如,它们包含环境暴露、过敏、正在进行或新出现的自身免疫疾病过程以及对免疫调节治疗的反应的证据。近年来,出现了高度多重化的方法来全面研究抗体结合特异性,这些方法成为重要的分子工具。在这里,我们提供了一个详细的方案,用于执行“噬菌体免疫沉淀测序”(PhIP-Seq),这是一种分析抗体库结合特异性的高通量、低成本的强大方法。该方法使用寡核苷酸文库合成(OLS)来编码蛋白质规模的肽文库,用于在噬菌体上展示。然后,使用个体的抗体对这些文库进行免疫沉淀,然后通过高通量 DNA 测序进行后续分析。我们已经使用 PhIP-Seq 来鉴定与自身免疫性疾病相关的新型自身抗原,来描述广谱中和 HIV 抗体的自身反应性,以及在一项针对数百种人类病毒暴露的大型国际横断面研究中。与替代的基于阵列的技术相比,PhIP-Seq 在样本通量和分析成本方面具有更大的可扩展性。不需要克隆和表达重组蛋白(与蛋白质微阵列相比),并且肽长度仅受 DNA 合成化学的限制(高达 90-aa(氨基酸)肽,而不是合成肽阵列的典型 8 到 12-aa 长度限制)。然而,与蛋白质微阵列相比,PhIP-Seq 文库缺乏不连续的表位和翻译后修饰。为了增加 PhIP-Seq 的可及性,我们提供了详细的说明,用于设计噬菌体展示的肽组文库、使用血清抗体进行免疫沉淀、基于深度测序的肽丰度测量以及统计确定反映抗体-肽相互作用的肽富集。一旦构建了文库,就可以在一周内获得 PhIP-Seq 数据进行分析。