Machine Learning Group, Technische Universität Berlin, 10587, Berlin, Germany.
Fritz-Haber-Institut der Max-Planck-Gesellschaft, 14195, Berlin, Germany.
Nat Commun. 2018 Sep 24;9(1):3887. doi: 10.1038/s41467-018-06169-2.
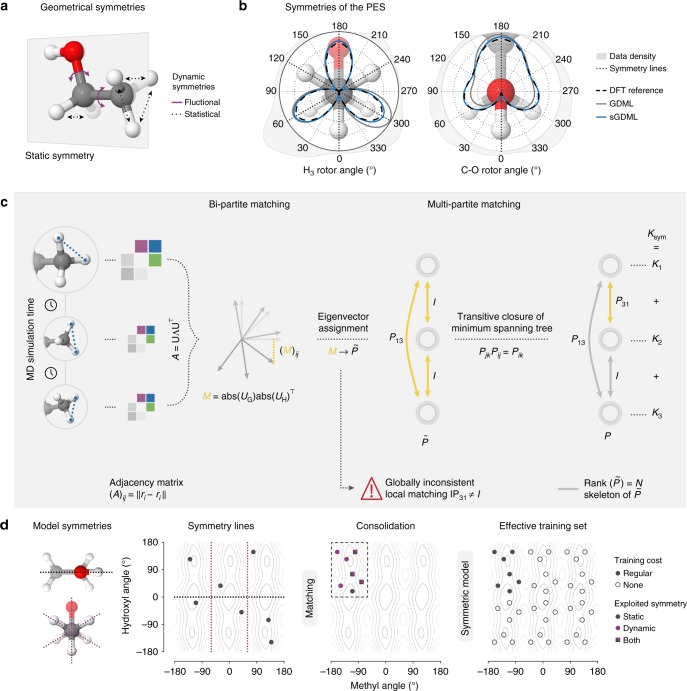
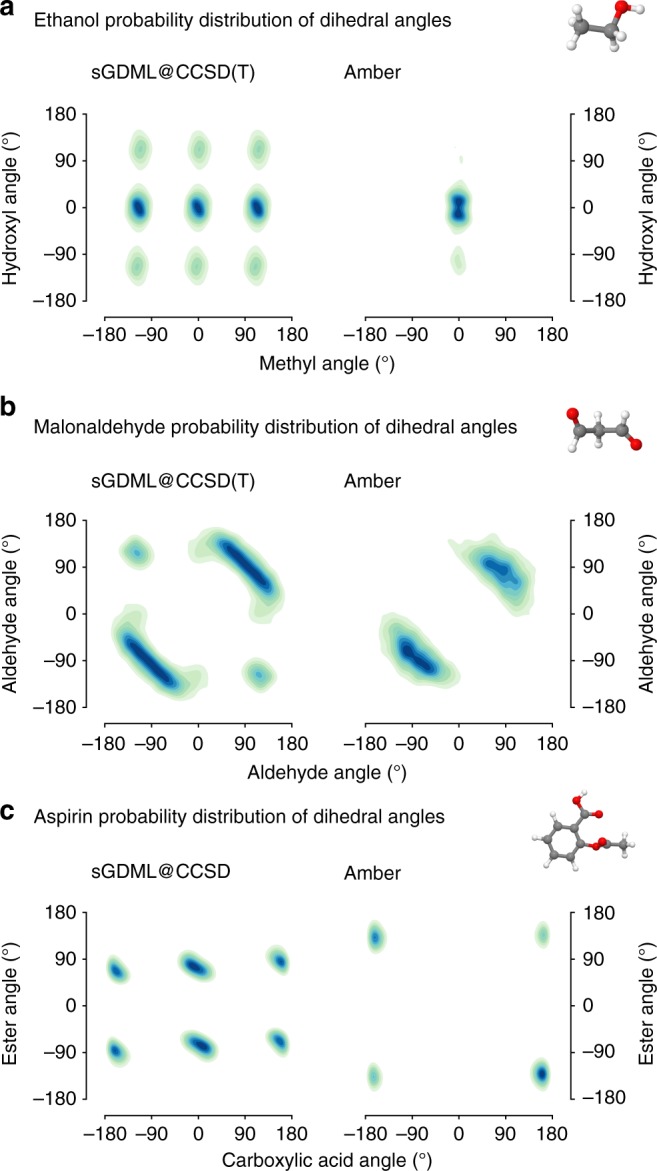
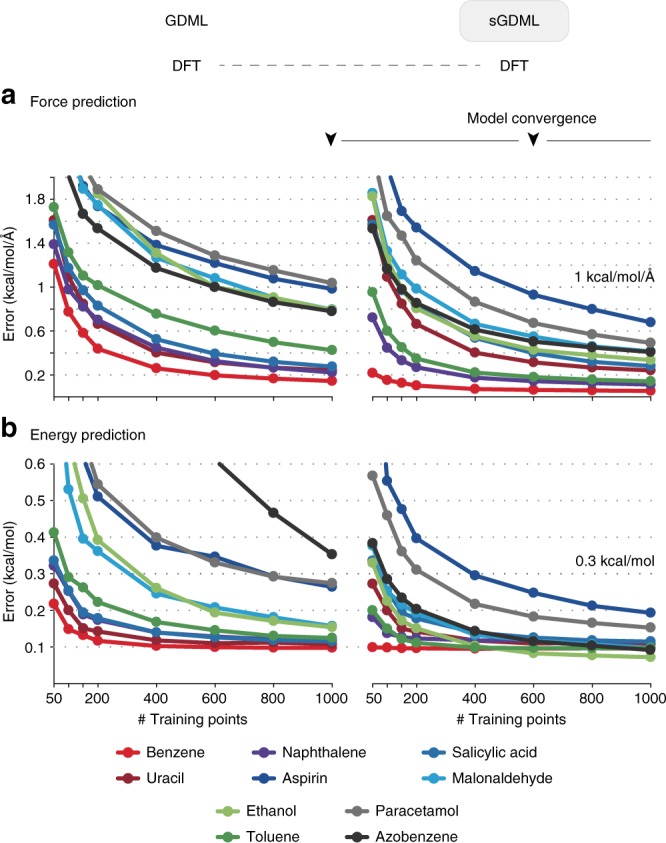
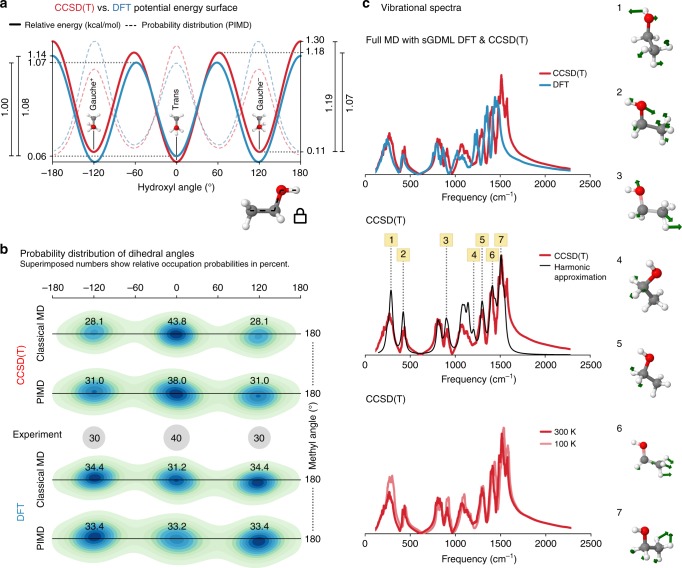
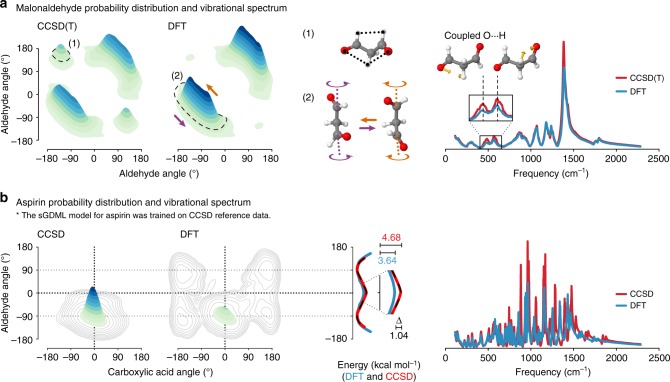
Molecular dynamics (MD) simulations employing classical force fields constitute the cornerstone of contemporary atomistic modeling in chemistry, biology, and materials science. However, the predictive power of these simulations is only as good as the underlying interatomic potential. Classical potentials often fail to faithfully capture key quantum effects in molecules and materials. Here we enable the direct construction of flexible molecular force fields from high-level ab initio calculations by incorporating spatial and temporal physical symmetries into a gradient-domain machine learning (sGDML) model in an automatic data-driven way. The developed sGDML approach faithfully reproduces global force fields at quantum-chemical CCSD(T) level of accuracy and allows converged molecular dynamics simulations with fully quantized electrons and nuclei. We present MD simulations, for flexible molecules with up to a few dozen atoms and provide insights into the dynamical behavior of these molecules. Our approach provides the key missing ingredient for achieving spectroscopic accuracy in molecular simulations.
分子动力学(MD)模拟采用经典力场,构成了化学、生物学和材料科学中当代原子建模的基石。然而,这些模拟的预测能力取决于潜在的原子间势。经典势能往往无法真实捕捉分子和材料中的关键量子效应。在这里,我们通过将时空物理对称性自动纳入梯度域机器学习(sGDML)模型,实现了从高级从头算计算中直接构建灵活分子力场的方法。所开发的 sGDML 方法忠实地再现了量子化学 CCSD(T)精度级别的全局力场,并允许使用完全量子化的电子和核进行收敛的分子动力学模拟。我们展示了多达几十个原子的灵活分子的 MD 模拟,并提供了这些分子动力学行为的深入见解。我们的方法为实现分子模拟的光谱精度提供了关键的缺失要素。