Department of Chemistry, Graduate School of Science, The University of Tokyo, Tokyo 113-0033, Japan.
Department of Chemistry, Graduate School of Science, The University of Tokyo, Tokyo 113-0033, Japan;
Proc Natl Acad Sci U S A. 2018 Oct 23;115(43):10959-10964. doi: 10.1073/pnas.1809901115. Epub 2018 Oct 9.
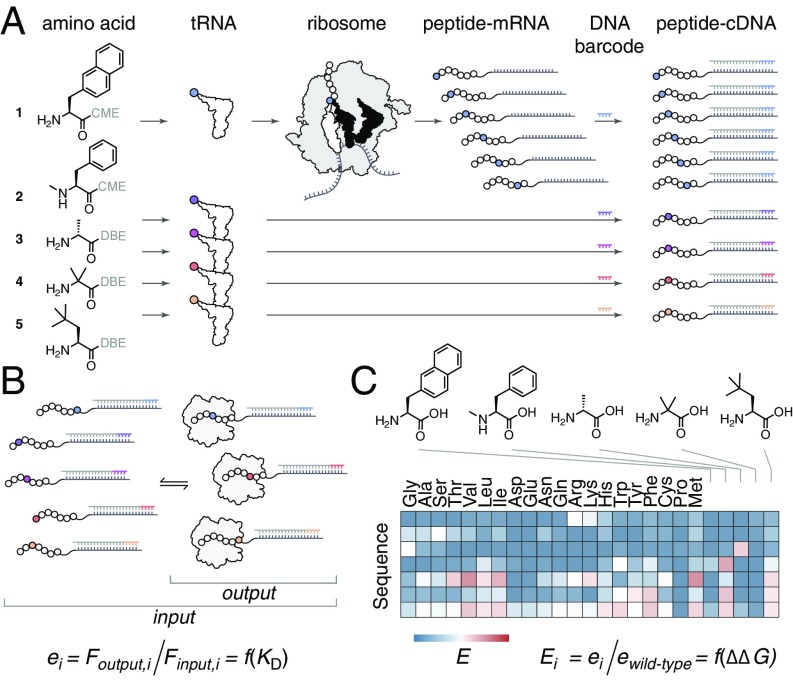
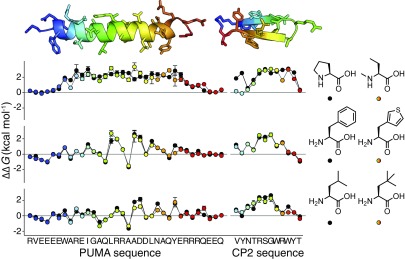
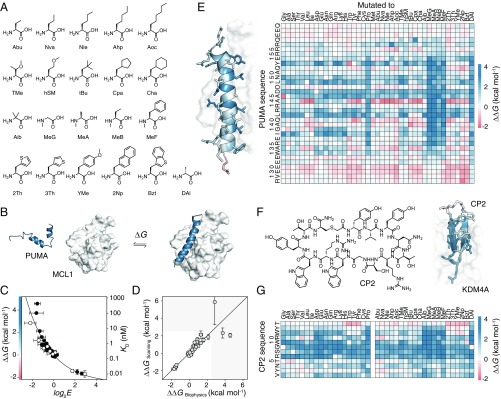
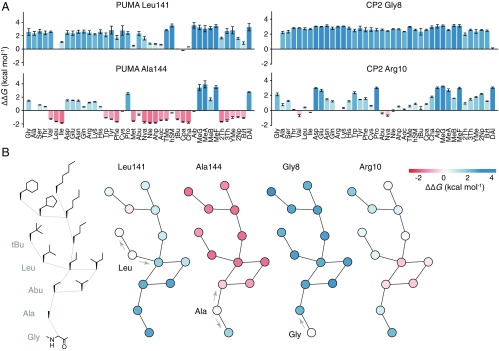
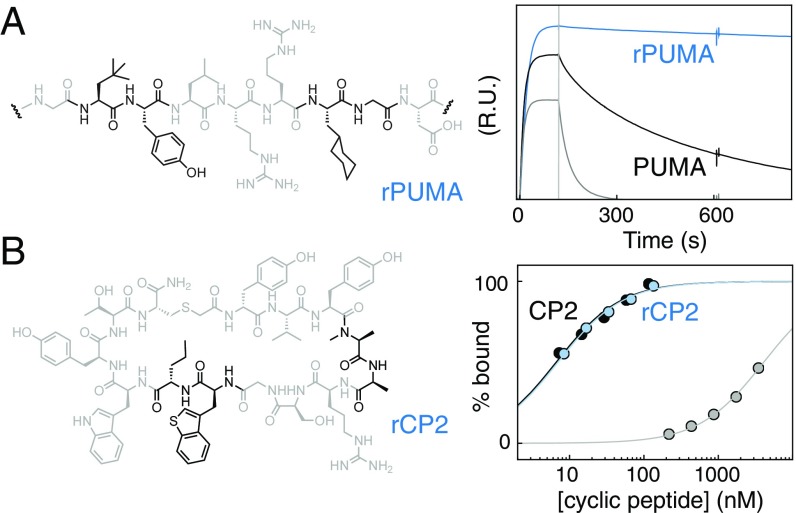
High-resolution structure-activity analysis of polypeptides requires amino acid structures that are not present in the universal genetic code. Examination of peptide and protein interactions with this resolution has been limited by the need to individually synthesize and test peptides containing nonproteinogenic amino acids. We describe a method to scan entire peptide sequences with multiple nonproteinogenic amino acids and, in parallel, determine the thermodynamics of binding to a partner protein. By coupling genetic code reprogramming to deep mutational scanning, any number of amino acids can be exhaustively substituted into peptides, and single experiments can return all free energy changes of binding. We validate this approach by scanning two model protein-binding peptides with 21 diverse nonproteinogenic amino acids. Dense structure-activity maps were produced at the resolution of single aliphatic atom insertions and deletions. This permits rapid interrogation of interaction interfaces, as well as optimization of affinity, fine-tuning of physical properties, and systematic assessment of nonproteinogenic amino acids in binding and folding.
要进行多肽的高分辨率结构-活性分析,就需要用到不存在于通用遗传密码中的氨基酸结构。由于需要逐个合成和测试含有非天然氨基酸的肽,因此对这种分辨率下的肽和蛋白质相互作用的研究一直受到限制。我们描述了一种方法,可以用多个非天然氨基酸对整个肽序列进行扫描,并同时确定与伴侣蛋白结合的热力学性质。通过将遗传密码重编程与深度突变扫描相结合,可以将任意数量的氨基酸彻底取代到肽中,并且单个实验可以返回所有结合的自由能变化。我们通过用 21 种不同的非天然氨基酸扫描两个模型蛋白结合肽来验证这种方法。以单个脂肪族原子插入和缺失的分辨率生成了密集的结构-活性图谱。这允许快速探究相互作用界面,以及优化亲和力、微调物理性质,并系统地评估结合和折叠中的非天然氨基酸。