Neuroscience Laboratory, The Cyprus Institute of Neurology and Genetics and Cyprus School of Molecular Medicine, Nicosia, Cyprus.
Department of Life and Health Sciences, School of Sciences and Engineering, University of Nicosia, Nicosia, Cyprus.
Glia. 2018 Dec;66(12):2589-2603. doi: 10.1002/glia.23513. Epub 2018 Oct 16.
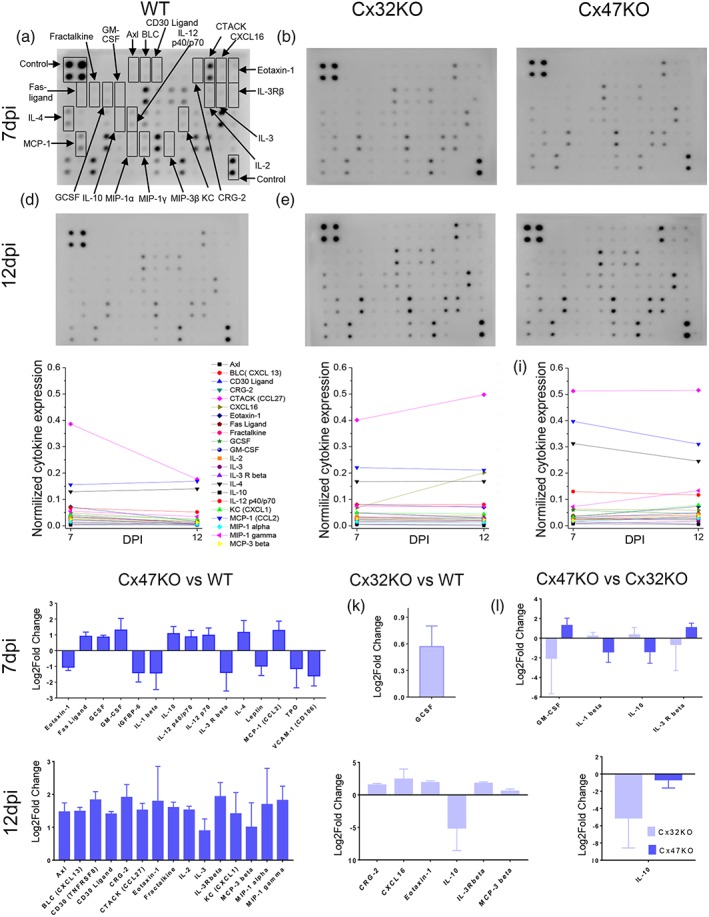
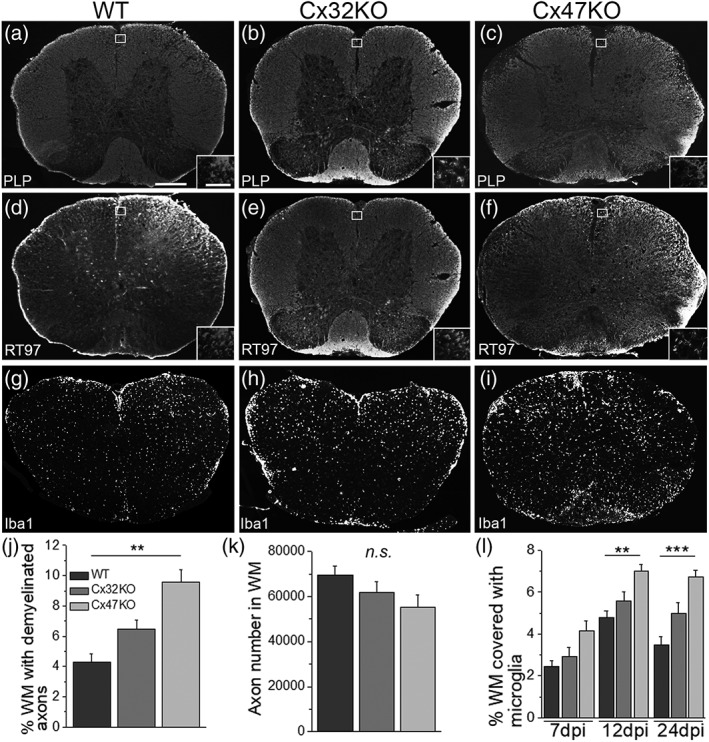
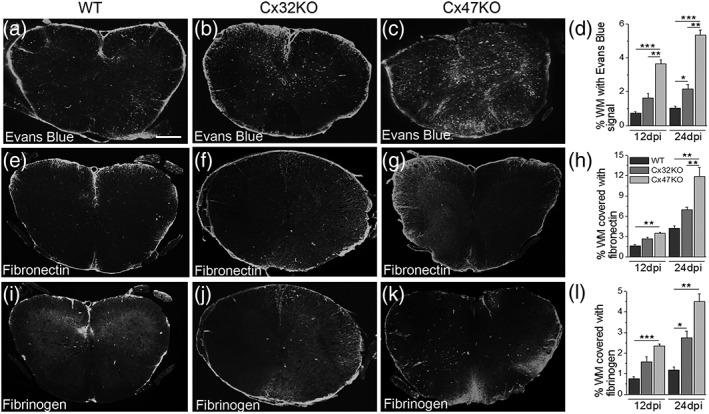
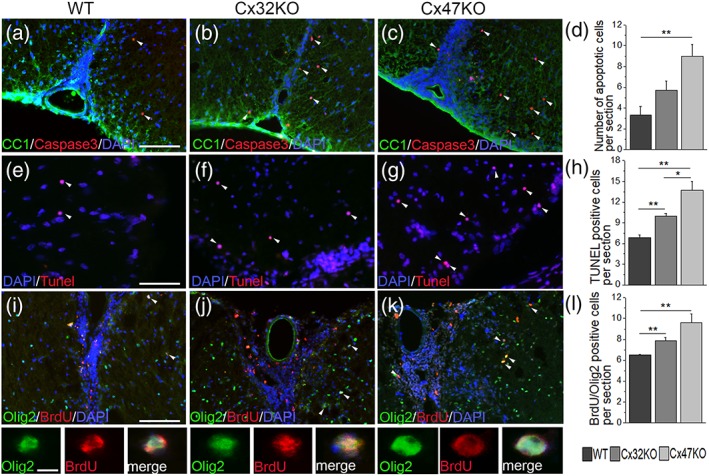
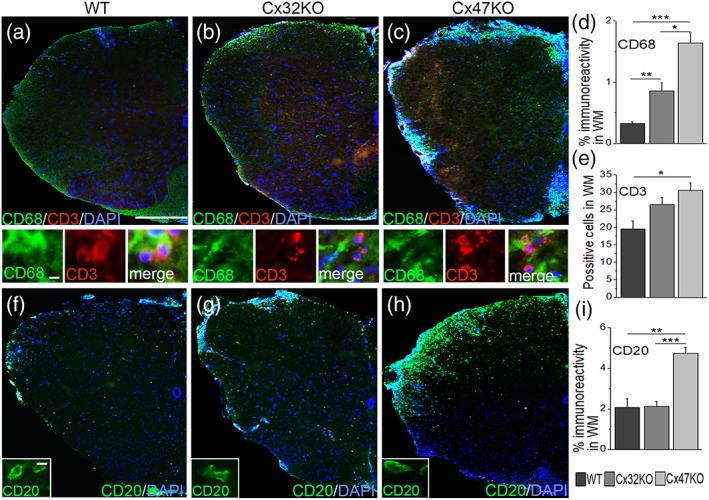
Gap junctions (GJs) coupling oligodendrocytes to astrocytes and to other oligodendrocytes are formed mainly by connexin47 (Cx47) and a smaller portion by connexin32 (Cx32). Mutations in both connexins cause inherited demyelinating disorders, but their expression is also disrupted in multiple sclerosis (MS). To clarify whether the loss of either Cx47 or Cx32 could modify the outcome of inflammation and myelin loss, we induced experimental autoimmune encephalomyelitis (EAE) in fully backcrossed Cx32 knockout (KO) and Cx47KO mice and compared their outcome with wild type (WT, C57BI/6 N) mice. Cx47KO EAE mice developed the most severe phenotype assessed by clinical scores and behavioral testing, followed by Cx32KO and WT mice. Cx47KO more than Cx32KO EAE mice developed more microglial activation, myelin, and axonal loss than did WT mice. Oligodendrocyte apoptosis and precursor proliferation was also higher in Cx47KO than in Cx32KO or WT EAE mice. Similarly, blood-spinal cord barrier (BSCB) disruption and inflammatory infiltrates of macrophages, T- and B-cells were more severe in Cx47KO than either Cx32KO or WT EAE groups. Finally, expression profiling revealed that several proinflammatory cytokines were higher at the peak of inflammation in the Cx47KO mice and persisted at later stages of EAE in contrast to reduction of their levels in WT EAE mice. Thus, loss of oligodendrocyte GJs aggravates BSCB disruption and inflammatory myelin loss, likely due to dysregulation of proinflammatory cytokines. This mechanism may play an important role in MS brain with reduced connexin expression, as well as in patients with inherited mutations in oligodendrocyte connexins and secondary inflammation.
缝隙连接(GJ)将少突胶质细胞与星形胶质细胞和其他少突胶质细胞偶联,主要由连接蛋白 47(Cx47)形成,较小部分由连接蛋白 32(Cx32)形成。这两种连接蛋白的突变都会导致遗传性脱髓鞘疾病,但在多发性硬化症(MS)中它们的表达也会受到干扰。为了阐明缺失任何一种 Cx47 或 Cx32 是否会改变炎症和髓鞘丢失的结果,我们在完全回交的 Cx32 敲除(KO)和 Cx47KO 小鼠中诱导实验性自身免疫性脑脊髓炎(EAE),并将其结果与野生型(WT,C57BI/6 N)小鼠进行比较。Cx47KO EAE 小鼠的临床评分和行为测试评估的表型最严重,其次是 Cx32KO 和 WT 小鼠。Cx47KO EAE 小鼠比 WT 小鼠发生更多的小胶质细胞激活、髓鞘和轴突丢失。少突胶质细胞凋亡和前体细胞增殖在 Cx47KO 小鼠中也高于 Cx32KO 或 WT EAE 小鼠。同样,血脊髓屏障(BSCB)破坏和巨噬细胞、T 细胞和 B 细胞的炎症浸润在 Cx47KO 比 Cx32KO 或 WT EAE 组更严重。最后,表达谱分析显示,在 Cx47KO 小鼠炎症高峰期几种促炎细胞因子表达升高,并且在 EAE 的后期阶段持续存在,而 WT EAE 小鼠的这些细胞因子水平降低。因此,少突胶质细胞 GJ 的缺失加重了 BSCB 的破坏和炎症性髓鞘丢失,可能是由于促炎细胞因子的失调。这种机制可能在 MS 大脑中连接蛋白表达降低,以及在少突胶质细胞连接蛋白遗传突变和继发性炎症的患者中发挥重要作用。