Department of Bioinformatics, Institute of Molecular and Cell Biology, University of Tartu, Tartu, Estonia.
Institute of Technology, University of Tartu, Tartu, Estonia.
PLoS Comput Biol. 2018 Oct 22;14(10):e1006434. doi: 10.1371/journal.pcbi.1006434. eCollection 2018 Oct.
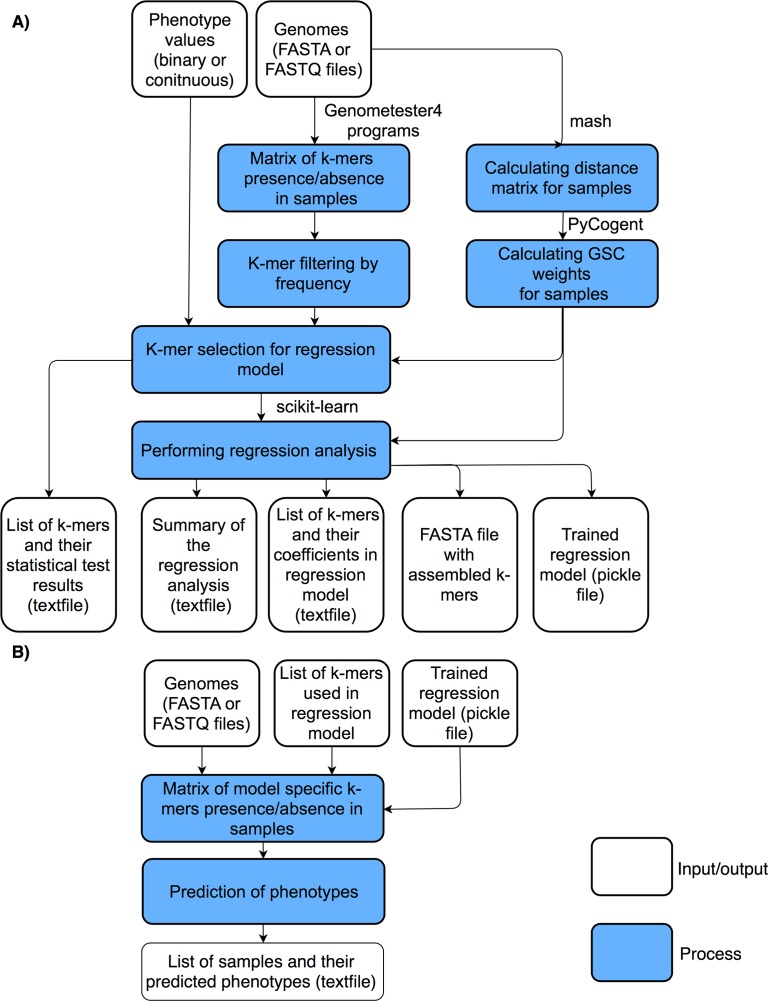
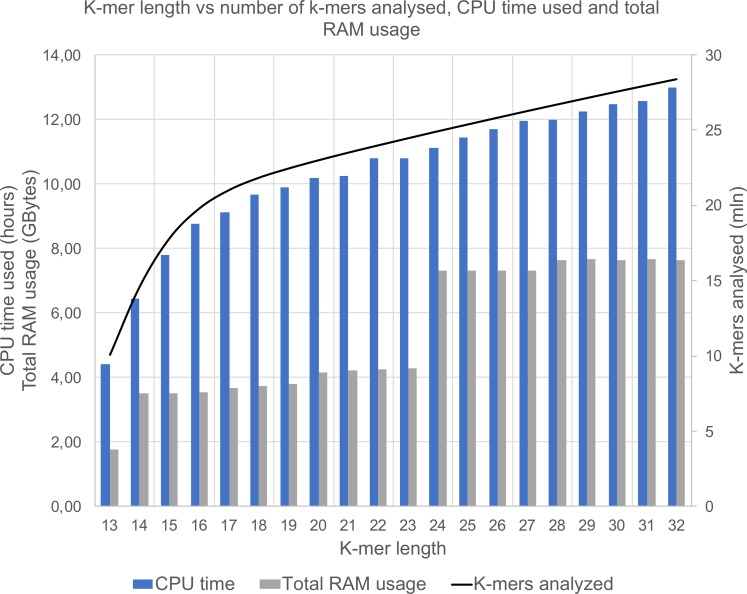
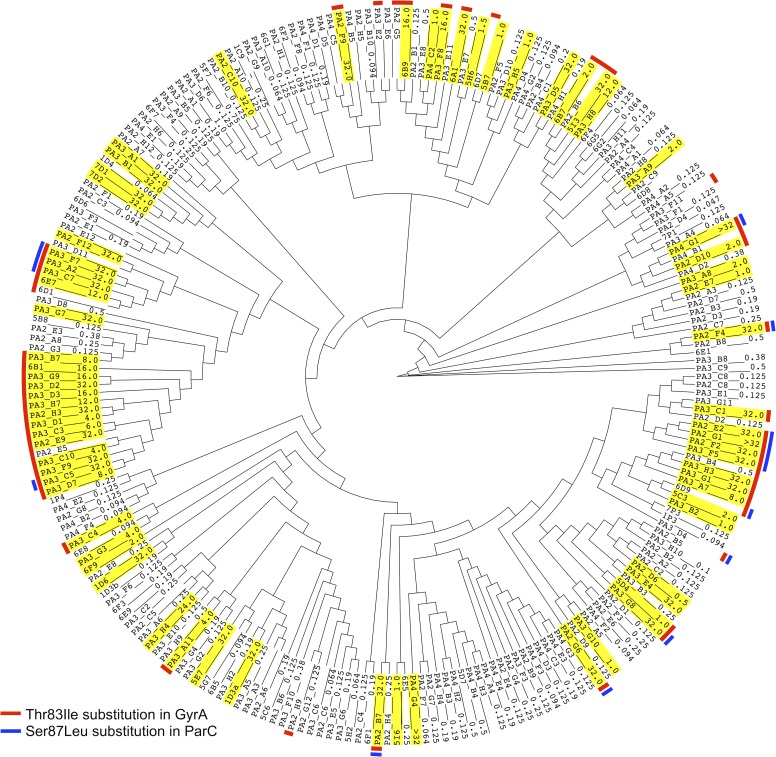
We have developed an easy-to-use and memory-efficient method called PhenotypeSeeker that (a) identifies phenotype-specific k-mers, (b) generates a k-mer-based statistical model for predicting a given phenotype and (c) predicts the phenotype from the sequencing data of a given bacterial isolate. The method was validated on 167 Klebsiella pneumoniae isolates (virulence), 200 Pseudomonas aeruginosa isolates (ciprofloxacin resistance) and 459 Clostridium difficile isolates (azithromycin resistance). The phenotype prediction models trained from these datasets obtained the F1-measure of 0.88 on the K. pneumoniae test set, 0.88 on the P. aeruginosa test set and 0.97 on the C. difficile test set. The F1-measures were the same for assembled sequences and raw sequencing data; however, building the model from assembled genomes is significantly faster. On these datasets, the model building on a mid-range Linux server takes approximately 3 to 5 hours per phenotype if assembled genomes are used and 10 hours per phenotype if raw sequencing data are used. The phenotype prediction from assembled genomes takes less than one second per isolate. Thus, PhenotypeSeeker should be well-suited for predicting phenotypes from large sequencing datasets. PhenotypeSeeker is implemented in Python programming language, is open-source software and is available at GitHub (https://github.com/bioinfo-ut/PhenotypeSeeker/).
我们开发了一种简单易用、内存效率高的方法,称为 PhenotypeSeeker,它 (a) 识别表型特异的 k-mers,(b) 生成基于 k-mer 的统计模型,用于预测给定的表型,(c) 从给定细菌分离物的测序数据预测表型。该方法在 167 株肺炎克雷伯菌分离株(毒力)、200 株铜绿假单胞菌分离株(环丙沙星耐药性)和 459 株艰难梭菌分离株(阿奇霉素耐药性)上进行了验证。从这些数据集训练的表型预测模型在肺炎克雷伯菌测试集中获得了 0.88 的 F1 度量,在铜绿假单胞菌测试集中获得了 0.88 的 F1 度量,在艰难梭菌测试集中获得了 0.97 的 F1 度量。F1 度量在组装序列和原始测序数据上是相同的;然而,从组装基因组构建模型要快得多。在这些数据集上,如果使用组装基因组,每个表型的模型构建大约需要 3 到 5 个小时,如果使用原始测序数据,每个表型需要 10 个小时。从组装基因组进行表型预测每个分离物不到 1 秒。因此,PhenotypeSeeker 应该非常适合从大型测序数据集预测表型。PhenotypeSeeker 是用 Python 编程语言实现的,是开源软件,可在 GitHub(https://github.com/bioinfo-ut/PhenotypeSeeker/)上获得。