Zhang Yanxi, Soni Saurabh, Krijger Theodorus L, Gordiichuk Pavlo, Qiu Xinkai, Ye Gang, Jonkman Harry T, Herrmann Andreas, Zojer Karin, Zojer Egbert, Chiechi Ryan C
Stratingh Institute for Chemistry , University of Groningen , Nijenborgh 4 , 9747 AG Groningen , The Netherlands.
Zernike Institute for Advanced Materials , Nijenborgh 4 , 9747 AG Groningen , The Netherlands.
J Am Chem Soc. 2018 Nov 7;140(44):15048-15055. doi: 10.1021/jacs.8b09793. Epub 2018 Oct 25.
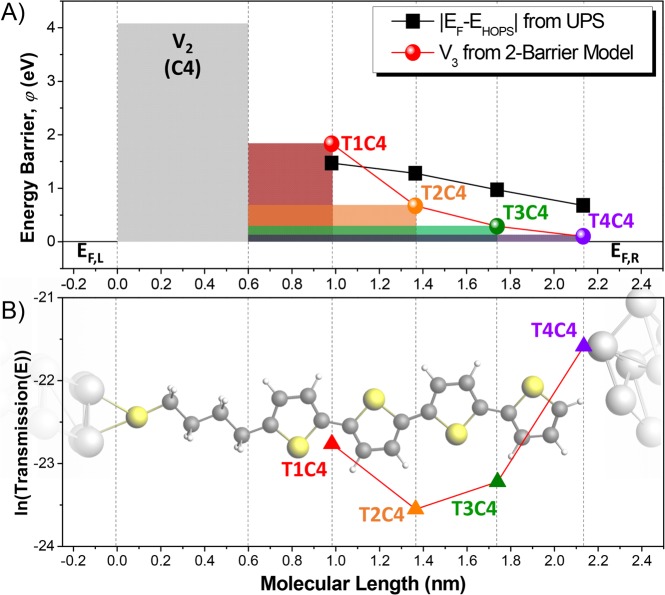
Molecular tunneling junctions should enable the tailoring of charge-transport at the quantum level through synthetic chemistry but are hindered by the dominance of the electrodes. We show that the frontier orbitals of molecules can be decoupled from the electrodes, preserving their relative energies in self-assembled monolayers even when a top-contact is applied. This decoupling leads to the remarkable observation of tunneling probabilities that increase with distance in a series of oligothiophenes, which we explain using a two-barrier tunneling model. This model is generalizable to any conjugated oligomers for which the frontier orbital gap can be determined and predicts that the molecular orbitals that dominate tunneling charge-transport can be positioned via molecular design rather than by domination of Fermi-level pinning arising from strong hybridization. The ability to preserve the electronic structure of molecules in tunneling junctions facilitates the application of well-established synthetic design rules to tailor the properties of molecular-electronic devices.
分子隧道结应能通过合成化学在量子水平上实现电荷传输的定制,但却受到电极主导作用的阻碍。我们表明,分子的前沿轨道可以与电极解耦,即使施加顶部接触,其相对能量在自组装单分子层中仍能得以保留。这种解耦导致了一个显著的观察结果:在一系列低聚噻吩中,隧穿概率随距离增加,我们用双势垒隧穿模型对此进行了解释。该模型可推广到任何能确定前沿轨道间隙的共轭低聚物,并预测主导隧穿电荷传输的分子轨道可通过分子设计来定位,而非由强杂化导致的费米能级钉扎主导。在隧道结中保留分子电子结构的能力有助于应用成熟的合成设计规则来定制分子电子器件的性能。