School of Chemistry , University of Southampton , Southampton , SO17 1BJ , U.K.
Computational and Systems Biology Program, Sloan Kettering Institute , Memorial Sloan Kettering Cancer Center , New York, New York 10065 , United States.
J Chem Theory Comput. 2018 Dec 11;14(12):6586-6597. doi: 10.1021/acs.jctc.8b00614. Epub 2018 Nov 19.
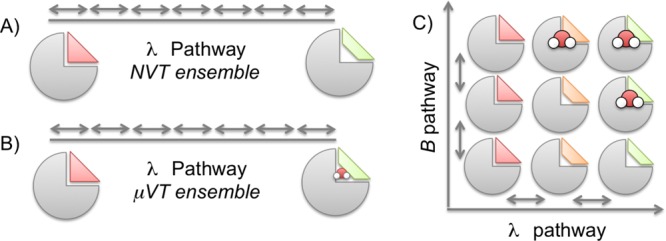
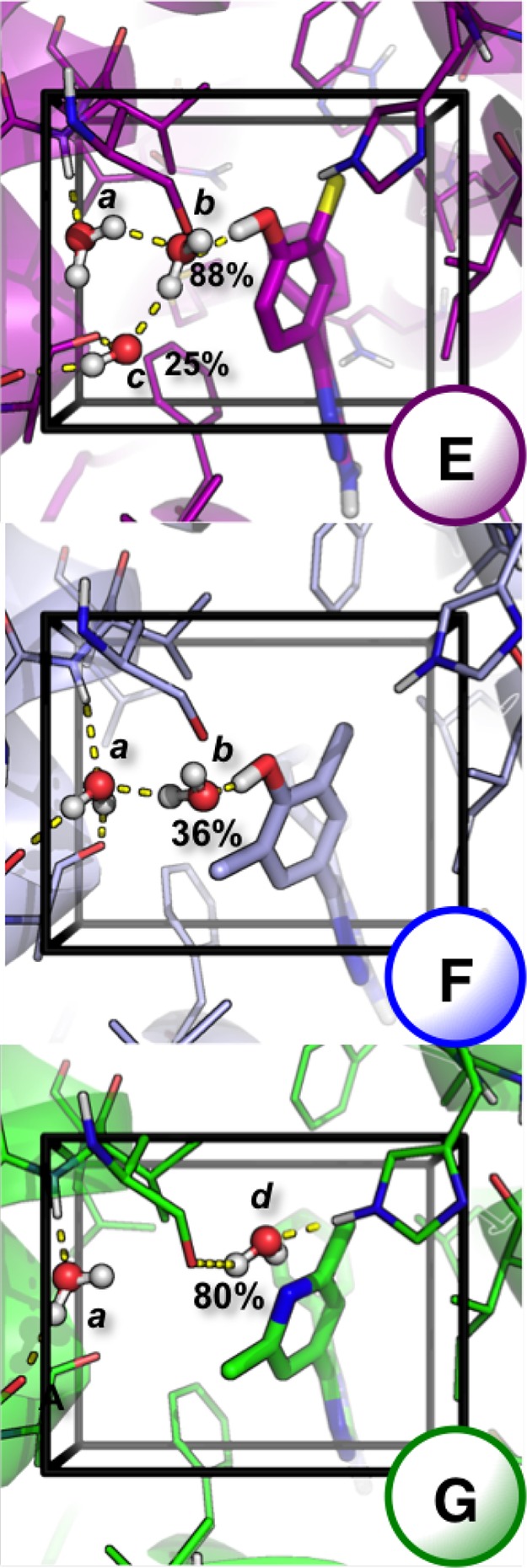
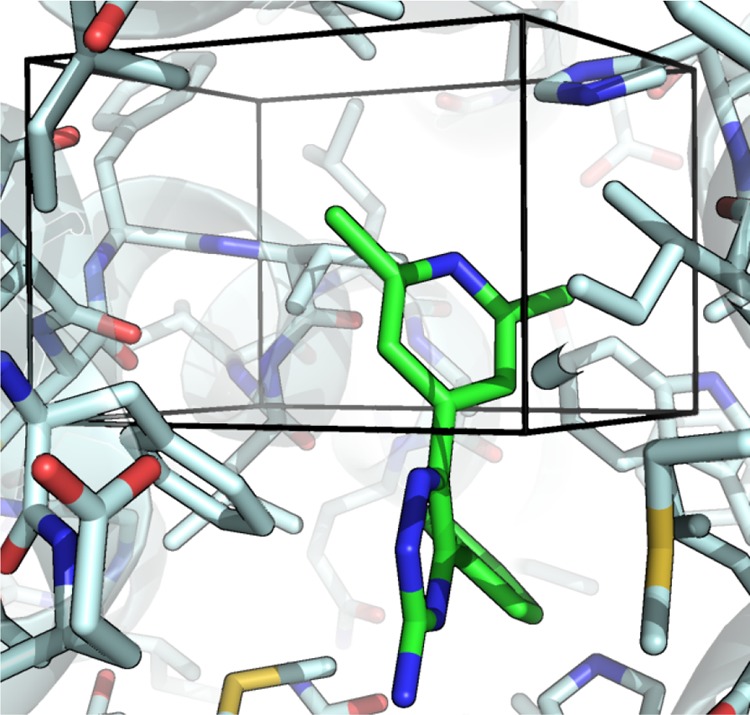
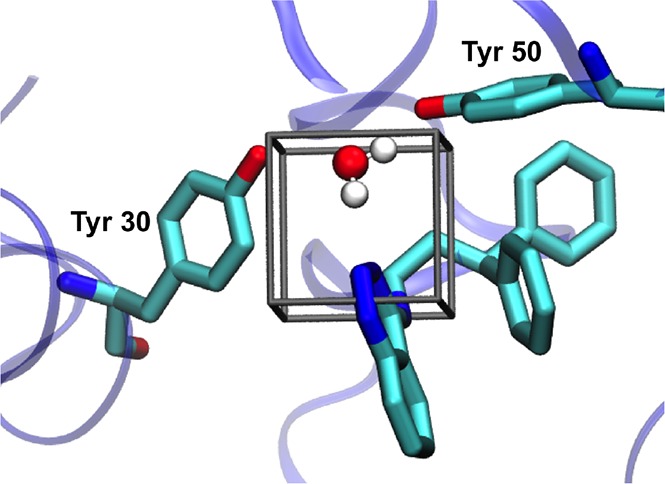
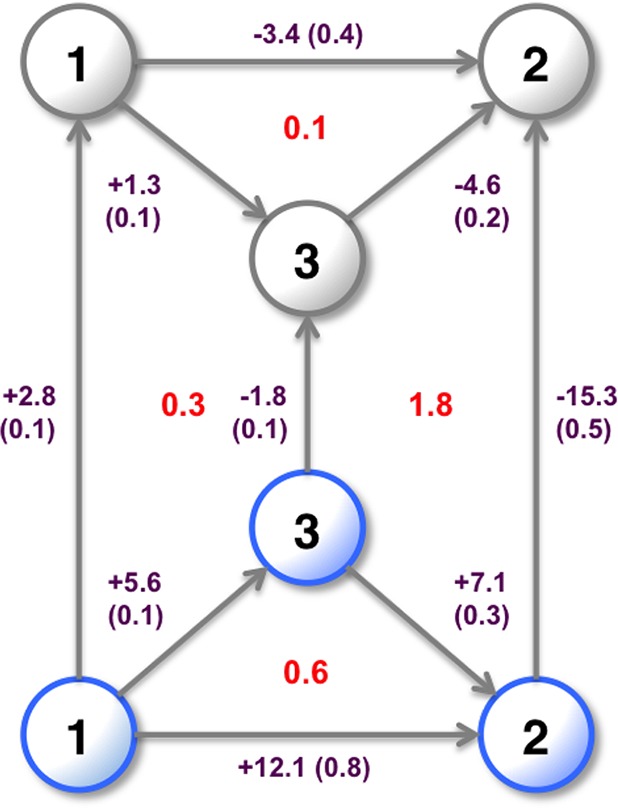
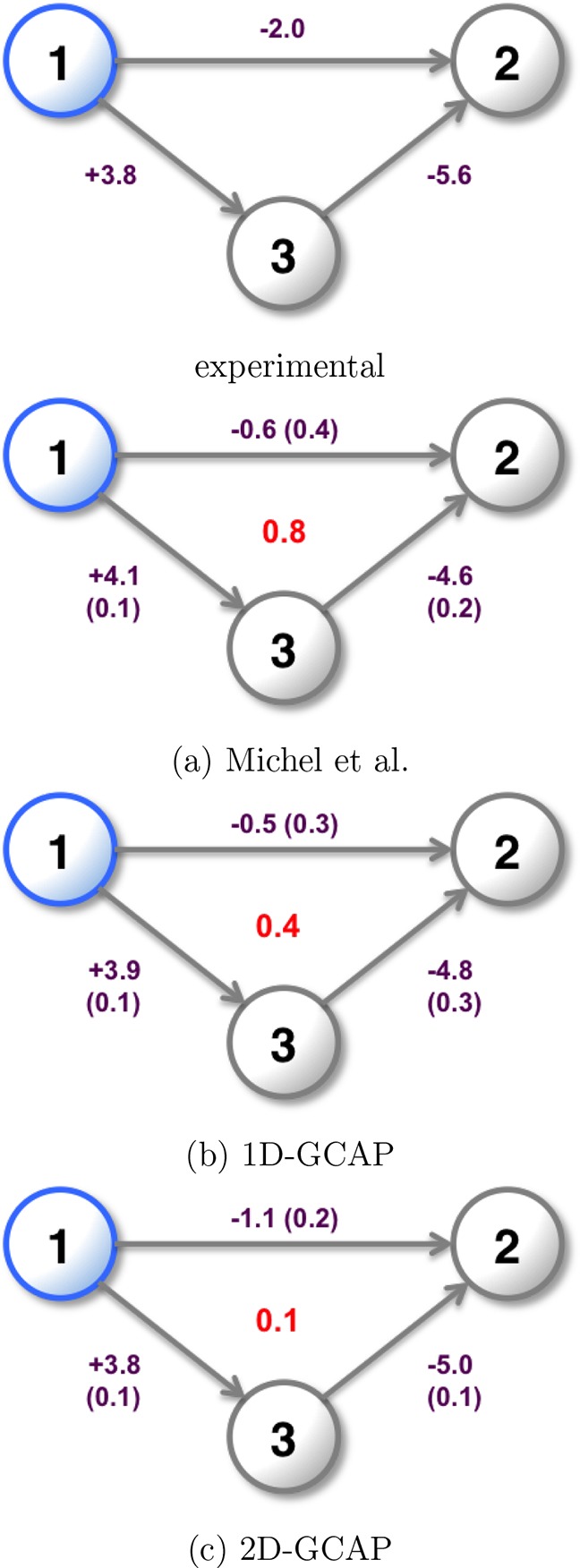
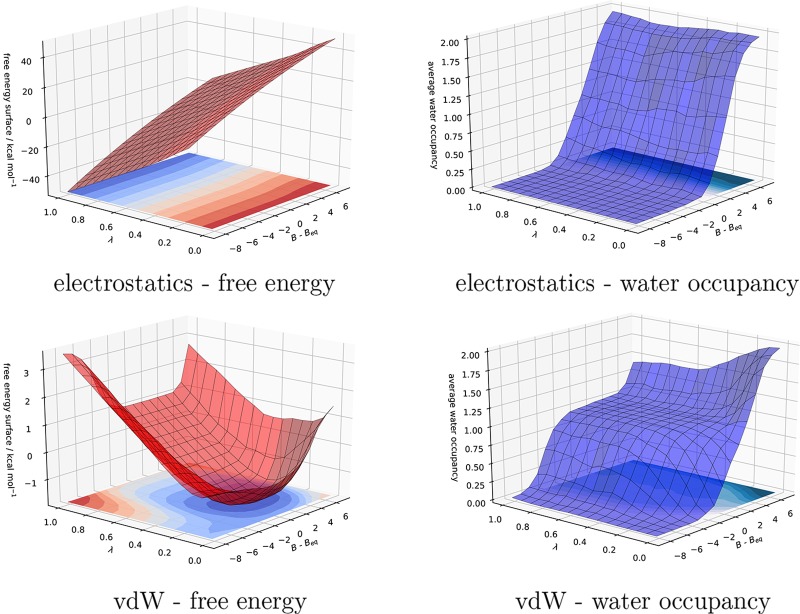
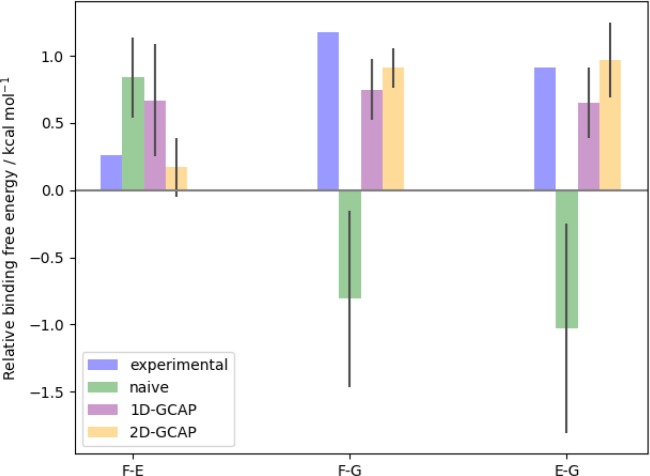
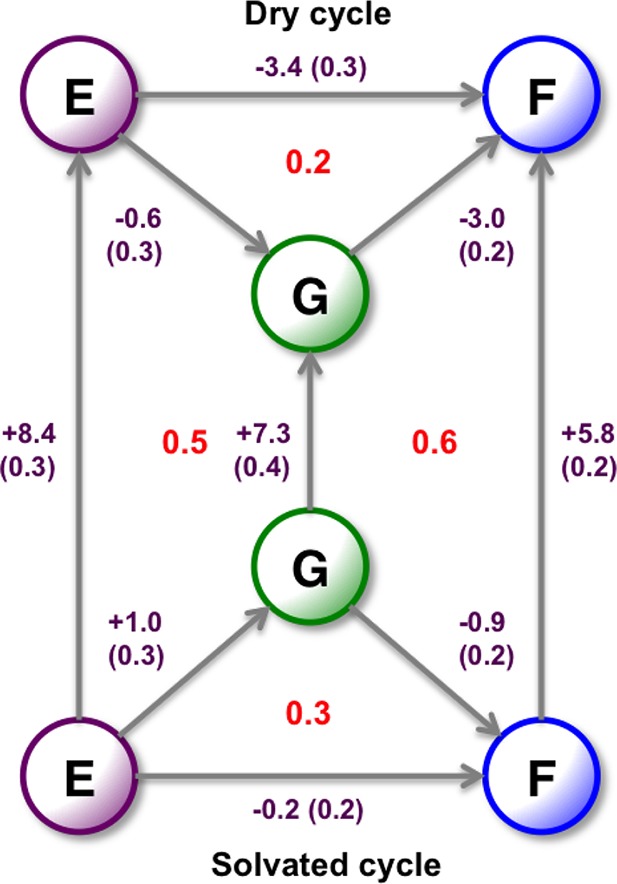
Computational methods to calculate ligand binding affinities are increasing in popularity, due to improvements in simulation algorithms, computational resources, and easy-to-use software. However, issues can arise in relative ligand binding free energy simulations if the ligands considered have different active site water networks, as simulations are typically performed with a predetermined number of water molecules (fixed N ensembles) in preassigned locations. If an alchemical perturbation is attempted where the change should result in a different active site water network, the water molecules may not be able to adapt appropriately within the time scales of the simulations-particularly if the active site is occluded. By combining the grand canonical ensemble (μVT) to sample active site water molecules, with conventional alchemical free energy methods, the water network is able to dynamically adapt to the changing ligand. We refer to this approach as grand canonical alchemical perturbation (GCAP). In this work we demonstrate GCAP for two systems; Scytalone Dehydratase (SD) and Adenosine A receptor. For both systems, GCAP is shown to perform well at reproducing experimental binding affinities. Calculating the relative binding affinities with a naı̈ve, conventional attempt to solvate the active site illustrates how poor results can be if proper consideration of water molecules in occluded pockets is neglected. GCAP results are shown to be consistent with time-consuming double decoupling simulations. In addition, by obtaining the free energy surface for ligand perturbations, as a function of both the free energy coupling parameter and water chemical potential, it is possible to directly deconvolute the binding energetics in terms of protein-ligand direct interactions and protein binding site hydration.
由于模拟算法、计算资源和易于使用的软件的改进,计算配体结合亲和力的计算方法越来越受欢迎。然而,如果所考虑的配体具有不同的活性位点水网络,那么在相对配体结合自由能模拟中可能会出现问题,因为模拟通常是在预定位置使用预定数量的水分子(固定 N 集合)进行的。如果尝试进行阿尔克里式扰动,其中变化应该导致不同的活性位点水网络,水分子可能无法在模拟的时间尺度内适当地适应 - 特别是如果活性位点被阻塞。通过将巨正则系综 (μVT) 与传统的阿尔克里自由能方法相结合,以采样活性位点水分子,水网络能够动态适应变化的配体。我们将这种方法称为巨正则阿尔克里式扰动 (GCAP)。在这项工作中,我们展示了 GCAP 在两个系统中的应用;苏氨酸脱水酶 (SD) 和腺苷 A 受体。对于这两个系统,GCAP 都能很好地重现实验结合亲和力。通过对活性位点进行简单的传统溶剂化尝试来计算相对结合亲和力,说明了如果忽略对被阻塞口袋中水分子的适当考虑,结果可能会很差。GCAP 的结果与耗时的双重去耦模拟一致。此外,通过获得作为自由能耦合参数和水化学势函数的配体扰动的自由能表面,可以直接解卷积配体与蛋白质的直接相互作用和蛋白质结合位点水合的结合能。