Department of Biochemistry and Molecular Biology, University of Chicago, Chicago, IL, 60637, USA.
Committee on Microbiology, University of Chicago, Chicago, IL, 60637, USA.
Nat Commun. 2018 Dec 17;9(1):5353. doi: 10.1038/s41467-018-07675-z.
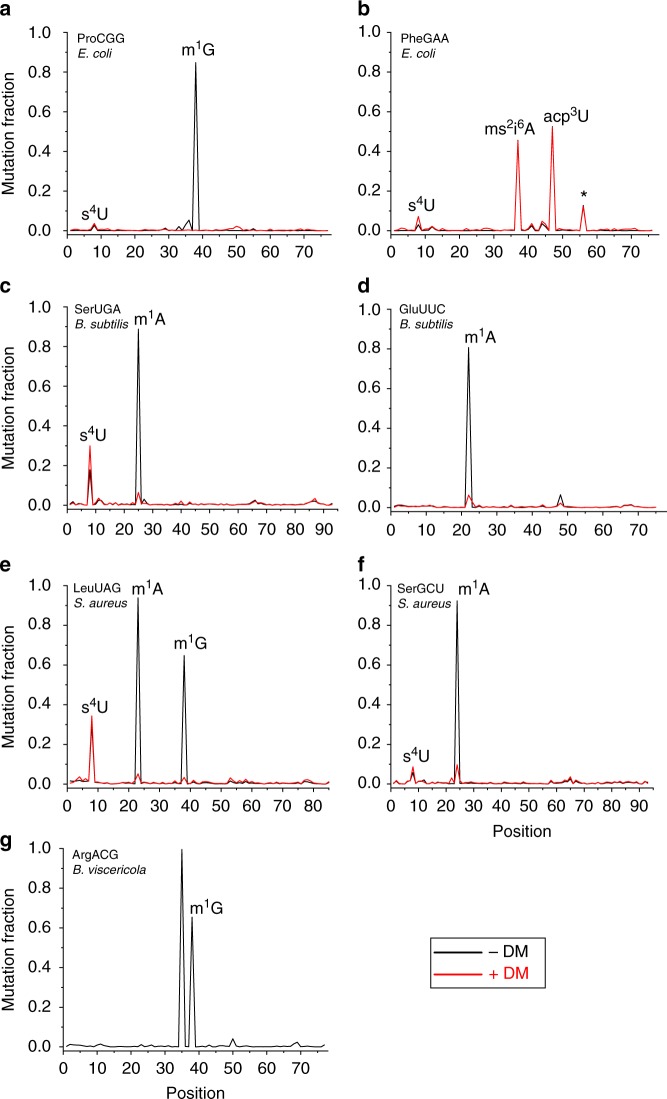
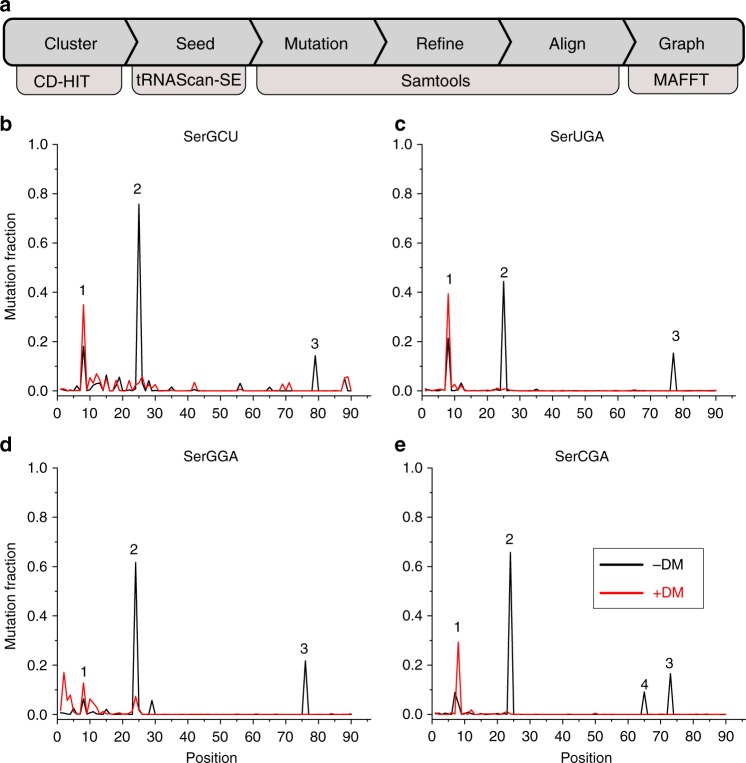
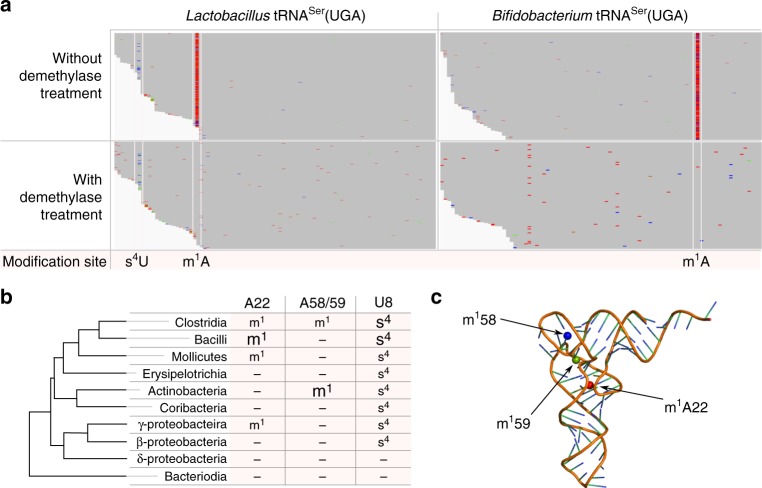
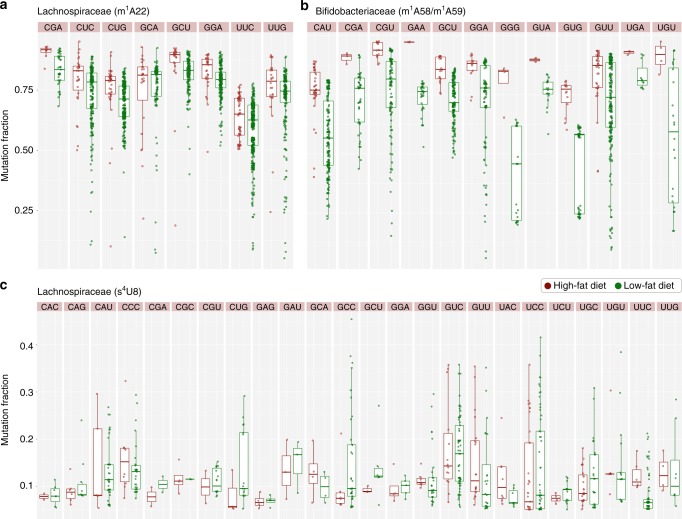
Advances in high-throughput sequencing have facilitated remarkable insights into the diversity and functioning of naturally occurring microbes; however, current sequencing strategies are insufficient to reveal physiological states of microbial communities associated with protein translation dynamics. Transfer RNAs (tRNAs) are core components of protein synthesis machinery, present in all living cells, and are phylogenetically tractable, which make them ideal targets to gain physiological insights into environmental microbes. Here we report a direct sequencing approach, tRNA-seq, and a software suite, tRNA-seq-tools, to recover sequences, abundance profiles, and post-transcriptional modifications of microbial tRNA transcripts. Our analysis of cecal samples using tRNA-seq distinguishes high-fat- and low-fat-fed mice in a comparable fashion to 16S ribosomal RNA gene amplicons, and reveals taxon- and diet-dependent variations in tRNA modifications. Our results provide taxon-specific in situ insights into the dynamics of tRNA gene expression and post-transcriptional modifications within complex environmental microbiomes.
高通量测序技术的进步极大地促进了对自然存在的微生物多样性和功能的深入了解;然而,目前的测序策略还不足以揭示与蛋白质翻译动态相关的微生物群落的生理状态。转移 RNA(tRNA)是蛋白质合成机制的核心组成部分,存在于所有活细胞中,并且在系统发育上是可追踪的,这使得它们成为获得环境微生物生理见解的理想目标。在这里,我们报告了一种直接测序方法 tRNA-seq 和一套软件套件 tRNA-seq-tools,用于恢复微生物 tRNA 转录物的序列、丰度分布和转录后修饰。我们使用 tRNA-seq 对盲肠样本的分析以类似于 16S 核糖体 RNA 基因扩增子的方式区分高脂肪和低脂肪喂养的小鼠,并揭示了 tRNA 修饰的分类群和饮食依赖性变化。我们的结果为特定于分类群的原位提供了关于复杂环境微生物组中 tRNA 基因表达和转录后修饰动态的见解。