Tian Saisai, Meng Guofeng, Zhang Weidong
Department of Phytochemistry, School of Pharmacy, Second Military Medical University, Shanghai 200433, People's Republic of China,
Institute of Interdisciplinary Integrative Medicine Research, Shanghai University of Traditional Chinese Medicine, Shanghai, People's Republic of China,
Cancer Manag Res. 2018 Dec 20;11:131-142. doi: 10.2147/CMAR.S185875. eCollection 2019.
Transcriptional dysregulation is one of the most important features of cancer genesis and progression. Applying gene expression dysregulation information to predict the development of cancers is useful for cancer diagnosis. However, previous studies mainly focused on the relationship between a single gene and cancer. Prognostic prediction using combined gene models remains limited.
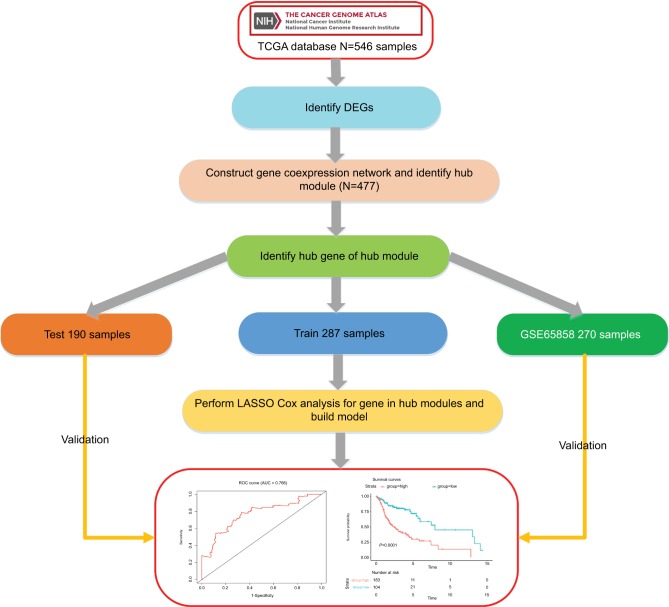
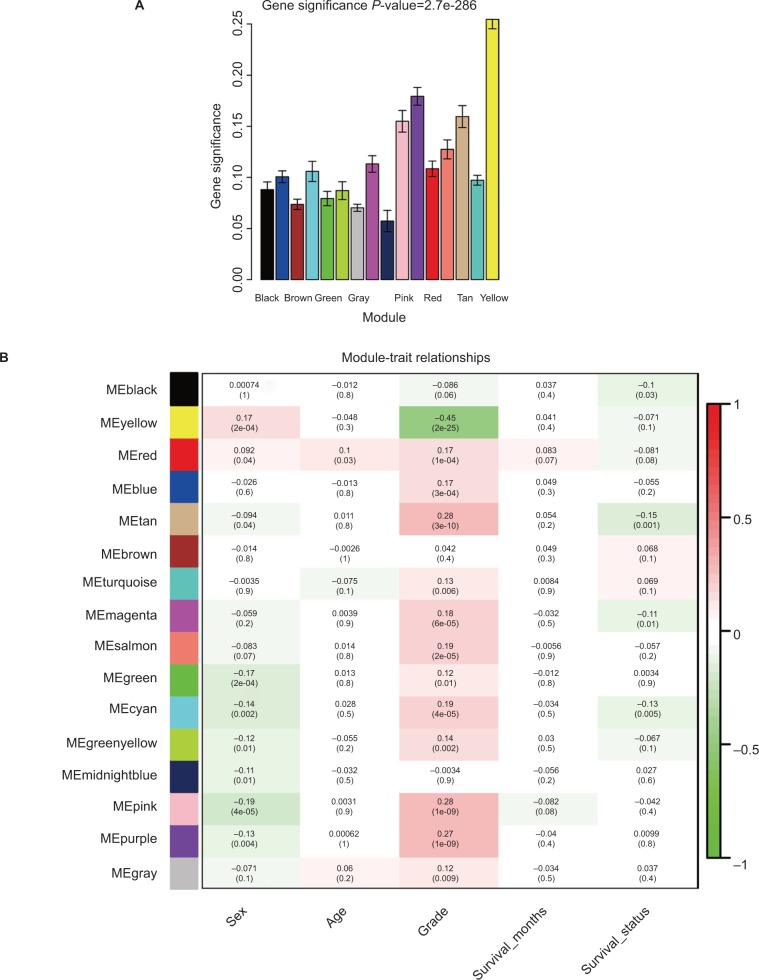
Gene expression profiles were downloaded from The Cancer Genome Atlas and the data sets were randomly divided into training data sets and test data sets. A six-gene signature associated with head and neck squamous cell carcinoma (HNSCC) and overall survival (OS) was identified according to a training cohort by using weighted gene correlation network analysis and least absolute shrinkage and selection operator Cox regression. The test data set and gene expression omnibus (GEO) data set were used to validate this signature.
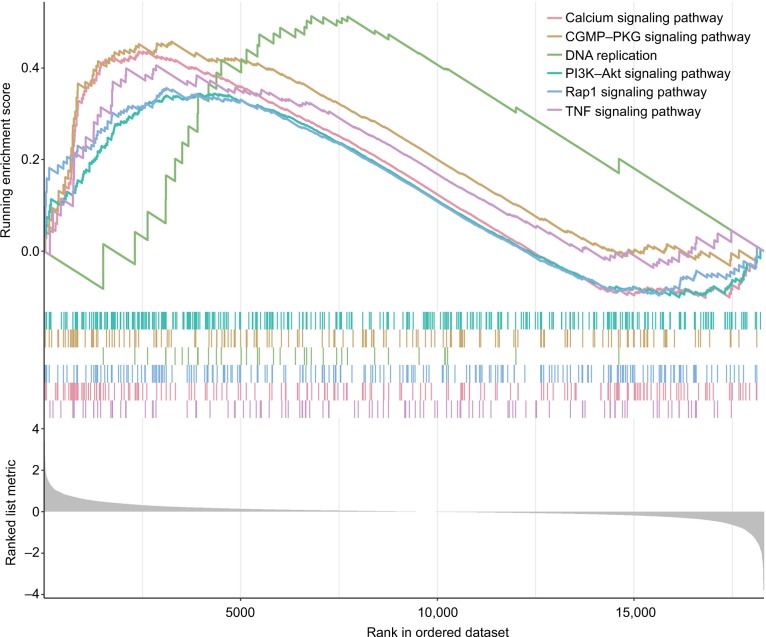
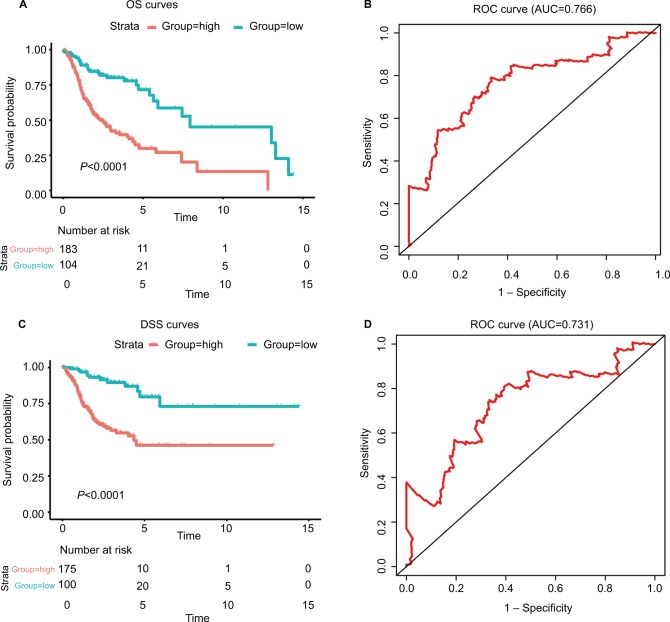
We identified six candidate genes, namely, FOXL2NB, PCOLCE2, SPINK6, ULBP2, KCNJ18, and RFPL1, and, using a six-gene model, predicted the risk of death of head and neck squamous cell carcinoma in The Cancer Genome Atlas. At a selected cutoff, patients were clustered into low- and high-risk groups. The OS curves of the two groups of patients had significant differences, and the time-dependent receiver operating characteristics of OS, disease-specific survival (DSS), and progression-free survival (PFS) were as high as 0.766, 0.731, and 0.623, respectively. Then, the test data set and the GEO data set were used to evaluate our model, and we found that the OS time in the high-risk group was significantly shorter than in the low-risk group in both data sets, and the receiver operating characteristics of test data set were 0.669, 0.675, and 0.614, respectively. Furthermore, univariate and multivariate Cox regression analyses showed that the risk score was independent of clinicopathological features.
The six-gene model could predict the OS of HNSCC patients and improve therapeutic decision-making.
转录失调是癌症发生和发展的最重要特征之一。应用基因表达失调信息预测癌症发展对癌症诊断很有用。然而,先前的研究主要集中在单个基因与癌症之间的关系。使用组合基因模型进行预后预测仍然有限。
从癌症基因组图谱下载基因表达谱,并将数据集随机分为训练数据集和测试数据集。通过加权基因共表达网络分析和最小绝对收缩和选择算子Cox回归,根据训练队列确定了一个与头颈部鳞状细胞癌(HNSCC)和总生存期(OS)相关的六基因特征。使用测试数据集和基因表达综合数据库(GEO)数据集验证该特征。
我们确定了六个候选基因,即FOXL2NB、PCOLCE2、SPINK6、ULBP2、KCNJ18和RFPL1,并使用六基因模型预测了癌症基因组图谱中头颈部鳞状细胞癌的死亡风险。在选定的临界值下,患者被分为低风险组和高风险组。两组患者的OS曲线有显著差异,OS、疾病特异性生存期(DSS)和无进展生存期(PFS)的时间依赖性受试者工作特征分别高达0.766、0.731和0.623。然后,使用测试数据集和GEO数据集评估我们的模型,我们发现在两个数据集中,高风险组的OS时间均显著短于低风险组,测试数据集的受试者工作特征分别为0.669、0.675和0.614。此外,单因素和多因素Cox回归分析表明,风险评分与临床病理特征无关。
六基因模型可以预测HNSCC患者的OS并改善治疗决策。