Zhao Shuang, Zhang Xuemei, Su Yanping, Chen Yilan, Liu Yang, Sun Meng, Qi Guohui
Department of Forestry, Agricultural University of Hebei, Baoding 071000, China.
Cotton Research Institute, Hebei Academy of Agriculture and Forestry Sciences, Shijiazhuang 050000, China.
Int J Genomics. 2018 Nov 28;2018:8931651. doi: 10.1155/2018/8931651. eCollection 2018.
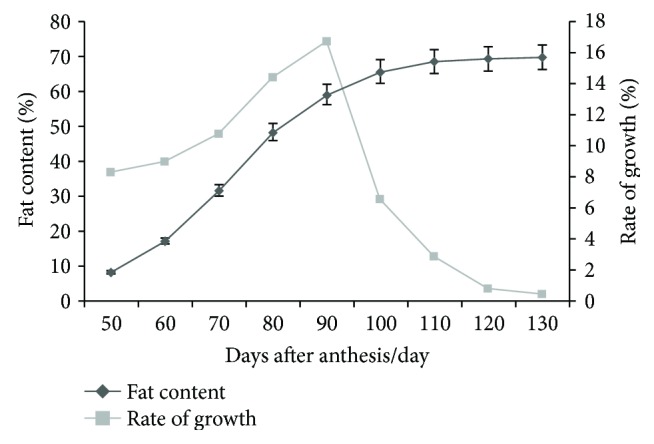
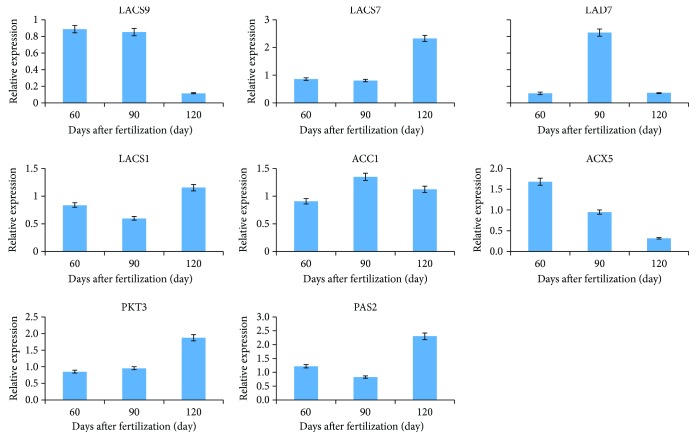
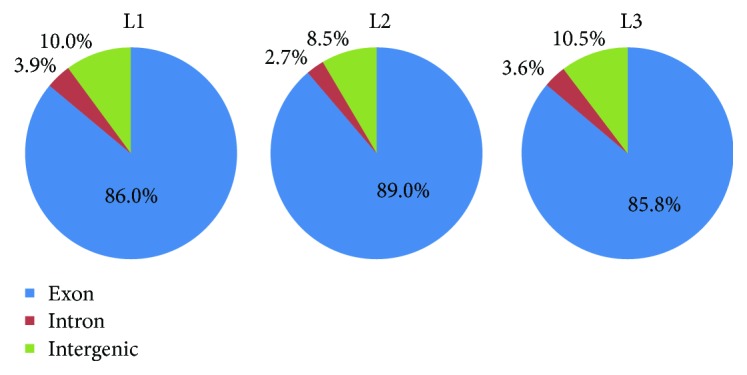
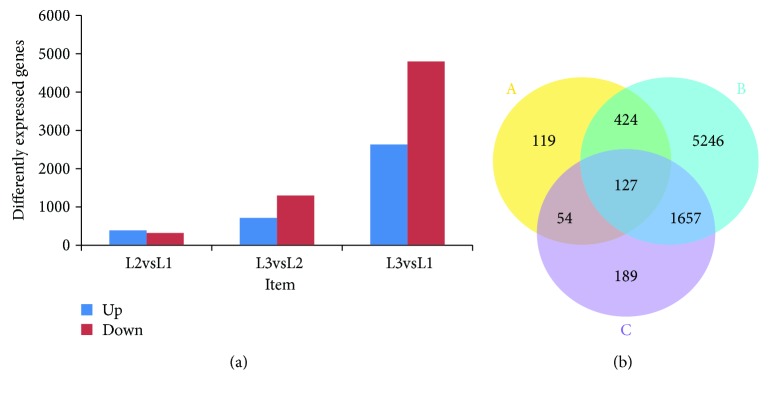
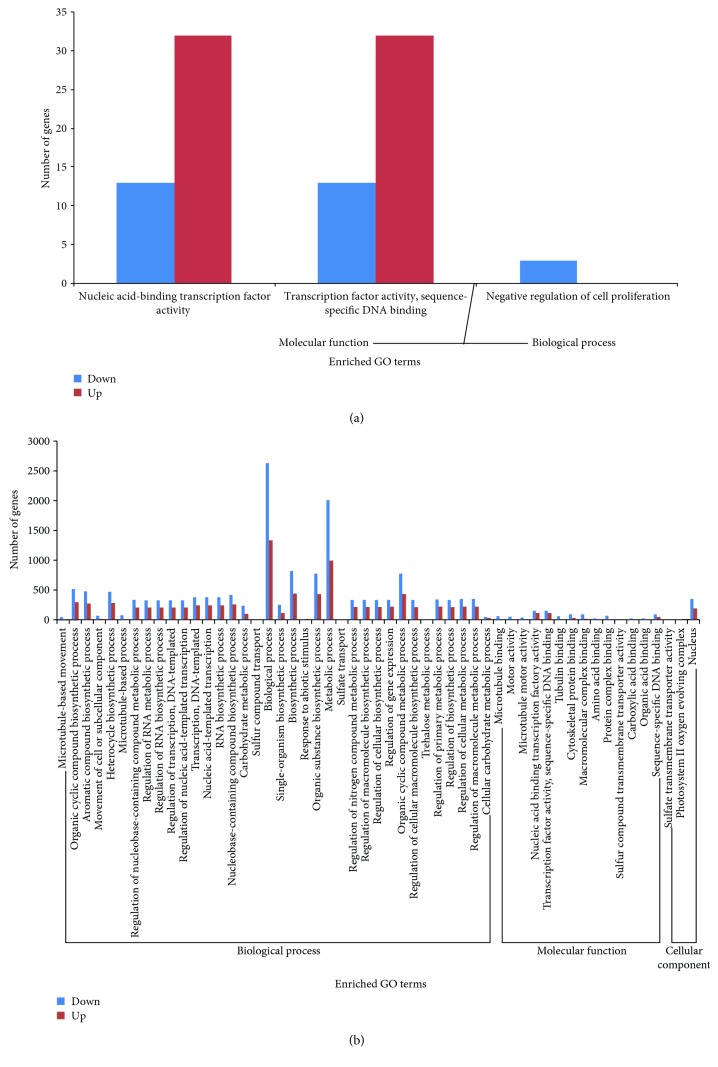
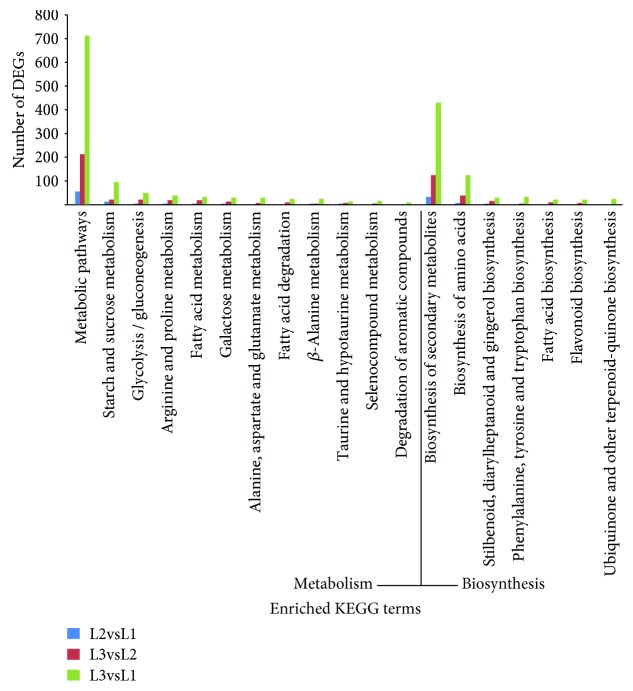
Walnut ( L.) is an important woody oilseed species cultivated throughout the world. In this study, comparative transcript profiling was performed using high-throughput RNA sequencing technology at the following three stages of walnut fat synthesis in the "Lvling" walnut cultivar: the initial developmental stage (L1), the fast developing stage (L2), and the last developing stage (L3). A total of 68.18 GB of data were obtained on the three developmental stages, and 92% to 94% of clean data were able to be located to the reference genome. Further comparisons of the transcripts in the three libraries revealed that 724, 2027, and 4817 genes were differentially expressed between the L2 and L1 (L2vsL1), L3 and L2 (L3vsL2), and L3 and L1 (L3vsL1) samples, respectively. Through the GO gene enrichment analysis, differentially expressed genes (DEGs) in L2vsL1, L3vsL2, and L3vsL1 were enriched into 3, 0, and 2 functional categories, respectively. According to the KEGG enrichment analysis, DEGs in L2vsL1, L3vsL2, and L3vsL1 were annotated into 77, 110, and 3717 taxonomic metabolic pathways in the KEGG database, respectively. Next, we analyzed expression levels of genes related to fat synthesis. Our results indicated that ACCase, LACS, and FAD7 were the key genes related to fat synthesis. The high-throughput transcriptome sequencing of walnut in different developmental stages has greatly enriched the current genomic available resources. The comparison of DEGs under different developmental stages identified a wealth of candidate genes involved in fat synthesis, which will facilitate further genetic improvement and molecular studies of the walnut.
核桃(Juglans regia L.)是一种在全球广泛种植的重要木本油料作物。在本研究中,利用高通量RNA测序技术,对“绿岭”核桃品种脂肪合成的三个阶段进行了比较转录组分析,这三个阶段分别为:初始发育阶段(L1)、快速发育阶段(L2)和最终发育阶段(L3)。在这三个发育阶段共获得了68.18 GB的数据,其中92%至94%的 clean数据能够定位到参考基因组上。进一步比较三个文库中的转录本发现,L2与L1(L2vsL1)、L3与L2(L3vsL2)以及L3与L1(L3vsL1)样本之间分别有724、2027和4817个基因差异表达。通过GO基因富集分析,L2vsL1、L3vsL2和L3vsL1中的差异表达基因(DEGs)分别富集到3、0和2个功能类别中。根据KEGG富集分析,L2vsL1、L3vsL2和L3vsL1中的DEGs分别注释到KEGG数据库中的77、110和3717个分类代谢途径中。接下来,我们分析了与脂肪合成相关基因的表达水平。结果表明,ACCase、LACS和FAD7是与脂肪合成相关的关键基因。不同发育阶段核桃的高通量转录组测序极大地丰富了当前可用的基因组资源。不同发育阶段DEGs的比较鉴定出了大量参与脂肪合成的候选基因,这将有助于核桃进一步的遗传改良和分子研究。