Institute of Biomedical Engineering, Department of Engineering Science, University of Oxford, Oxford, UK.
Oxford-Suzhou Centre for Advanced Research, Suzhou, China.
Bioinformatics. 2019 Sep 15;35(18):3240-3249. doi: 10.1093/bioinformatics/btz067.
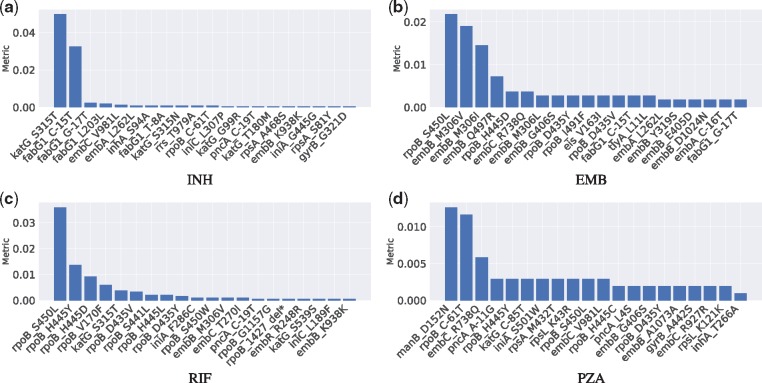
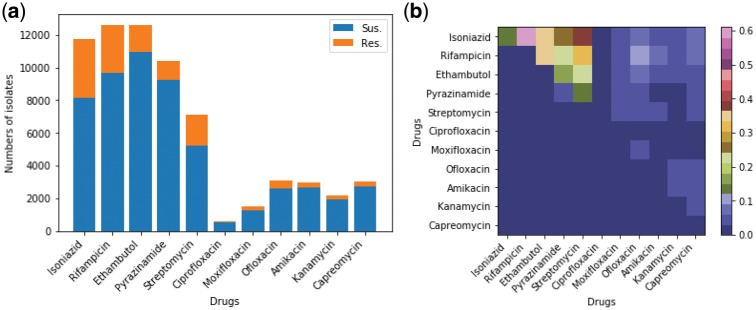
Resistance co-occurrence within first-line anti-tuberculosis (TB) drugs is a common phenomenon. Existing methods based on genetic data analysis of Mycobacterium tuberculosis (MTB) have been able to predict resistance of MTB to individual drugs, but have not considered the resistance co-occurrence and cannot capture latent structure of genomic data that corresponds to lineages.
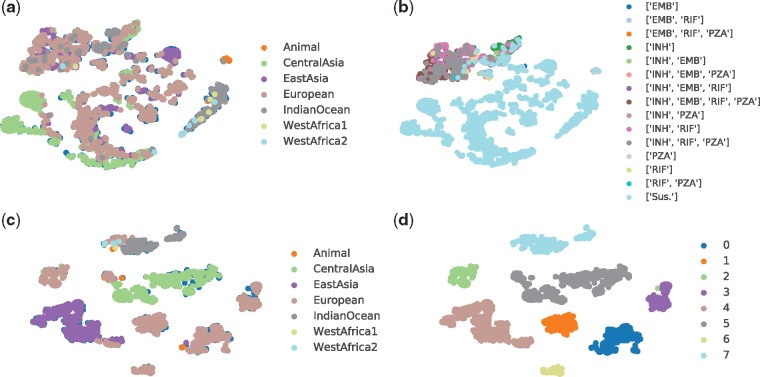
We used a large cohort of TB patients from 16 countries across six continents where whole-genome sequences for each isolate and associated phenotype to anti-TB drugs were obtained using drug susceptibility testing recommended by the World Health Organization. We then proposed an end-to-end multi-task model with deep denoising auto-encoder (DeepAMR) for multiple drug classification and developed DeepAMR_cluster, a clustering variant based on DeepAMR, for learning clusters in latent space of the data. The results showed that DeepAMR outperformed baseline model and four machine learning models with mean AUROC from 94.4% to 98.7% for predicting resistance to four first-line drugs [i.e. isoniazid (INH), ethambutol (EMB), rifampicin (RIF), pyrazinamide (PZA)], multi-drug resistant TB (MDR-TB) and pan-susceptible TB (PANS-TB: MTB that is susceptible to all four first-line anti-TB drugs). In the case of INH, EMB, PZA and MDR-TB, DeepAMR achieved its best mean sensitivity of 94.3%, 91.5%, 87.3% and 96.3%, respectively. While in the case of RIF and PANS-TB, it generated 94.2% and 92.2% sensitivity, which were lower than baseline model by 0.7% and 1.9%, respectively. t-SNE visualization shows that DeepAMR_cluster captures lineage-related clusters in the latent space.
The details of source code are provided at http://www.robots.ox.ac.uk/∼davidc/code.php.
Supplementary data are available at Bioinformatics online.
一线抗结核 (TB) 药物的耐药性共存是一种常见现象。现有的基于分枝杆菌 (MTB) 遗传数据分析的方法已经能够预测 MTB 对个别药物的耐药性,但没有考虑耐药性共存,也无法捕捉与谱系相对应的基因组数据的潜在结构。
我们使用了来自六大洲 16 个国家的大量结核病患者队列,这些患者的全基因组序列是使用世界卫生组织推荐的抗结核药物药敏试验获得的。然后,我们提出了一个端到端的多任务模型,该模型使用深度去噪自动编码器 (DeepAMR) 进行多种药物分类,并开发了 DeepAMR_cluster,这是一种基于 DeepAMR 的聚类变体,用于学习数据潜在空间中的聚类。结果表明,DeepAMR 优于基线模型和其他四个机器学习模型,其对四种一线药物(即异烟肼 (INH)、乙胺丁醇 (EMB)、利福平 (RIF)、吡嗪酰胺 (PZA))、耐多药结核病 (MDR-TB) 和全敏感结核病 (PANS-TB:对四种一线抗结核药物均敏感的 MTB) 的预测耐药性的平均 AUROC 从 94.4%到 98.7%不等。在 INH、EMB、PZA 和 MDR-TB 的情况下,DeepAMR 分别实现了其最佳的平均敏感性 94.3%、91.5%、87.3%和 96.3%。而在 RIF 和 PANS-TB 的情况下,它生成的敏感性分别为 94.2%和 92.2%,比基线模型低 0.7%和 1.9%。t-SNE 可视化显示,DeepAMR_cluster 在潜在空间中捕获了与谱系相关的聚类。
源代码的详细信息在 http://www.robots.ox.ac.uk/∼davidc/code.php 提供。
补充数据可在《生物信息学》在线获取。