College of Electrical Engineering and Automation, Anhui University, Hefei, 230601, Anhui, China.
Institute of Physical Science and Information Technology, School of Computer Science and Technology, Anhui University, 111 Jiulong Avenue, Hefei, 230601, China.
BMC Med Genomics. 2019 Jan 31;12(Suppl 1):12. doi: 10.1186/s12920-018-0455-6.
Although synonymous single nucleotide variants (sSNVs) do not alter the protein sequences, they have been shown to play an important role in human disease. Distinguishing pathogenic sSNVs from neutral ones is challenging because pathogenic sSNVs tend to have low prevalence. Although many methods have been developed for predicting the functional impact of single nucleotide variants, only a few have been specifically designed for identifying pathogenic sSNVs.
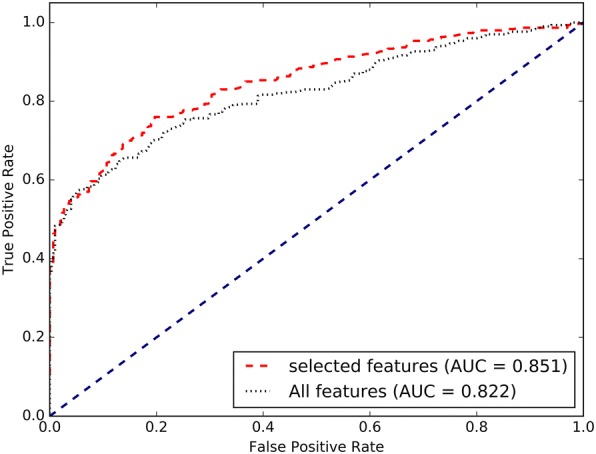
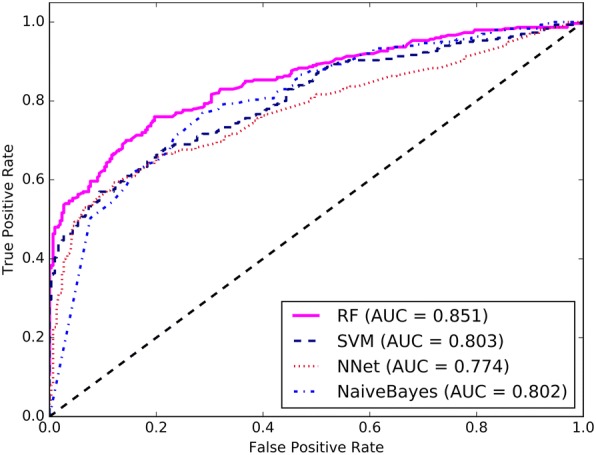
In this work, we describe a computational model, IDSV (Identification of Deleterious Synonymous Variants), which uses random forest (RF) to detect deleterious sSNVs in human genomes. We systematically investigate a total of 74 multifaceted features across seven categories: splicing, conservation, codon usage, sequence, pre-mRNA folding energy, translation efficiency, and function regions annotation features. Then, to remove redundant and irrelevant features and improve the prediction performance, feature selection is employed using the sequential backward selection method. Based on the optimized 10 features, a RF classifier is developed to identify deleterious sSNVs. The results on benchmark datasets show that IDSV outperforms other state-of-the-art methods in identifying sSNVs that are pathogenic.
We have developed an efficient feature-based prediction approach (IDSV) for deleterious sSNVs by using a wide variety of features. Among all the features, a compact and useful feature subset that has an important implication for identifying deleterious sSNVs is identified. Our results indicate that besides splicing and conservation features, a new translation efficiency feature is also an informative feature for identifying deleterious sSNVs. While the function regions annotation and sequence features are weakly informative, they may have the ability to discriminate deleterious sSNVs from benign ones when combined with other features. The data and source code are available on website http://bioinfo.ahu.edu.cn:8080/IDSV .
虽然同义单核苷酸变异(sSNV)不会改变蛋白质序列,但它们已被证明在人类疾病中发挥重要作用。区分致病性 sSNV 和中性 sSNV 具有挑战性,因为致病性 sSNV 往往患病率较低。尽管已经开发了许多用于预测单核苷酸变异功能影响的方法,但只有少数方法专门用于识别致病性 sSNV。
在这项工作中,我们描述了一种计算模型 IDSV(有害同义变异识别),它使用随机森林(RF)来检测人类基因组中的有害 sSNV。我们系统地研究了七个类别中的 74 个多方面特征:剪接、保守性、密码子使用、序列、前 mRNA 折叠能、翻译效率和功能区域注释特征。然后,使用逐步后向选择方法进行特征选择,以去除冗余和不相关的特征并提高预测性能。基于优化的 10 个特征,开发了 RF 分类器来识别有害 sSNV。在基准数据集上的结果表明,IDSV 在识别致病性 sSNV 方面优于其他最先进的方法。
我们通过使用各种特征开发了一种有效的基于特征的预测方法(IDSV)来预测有害 sSNV。在所有特征中,确定了一个紧凑且有用的特征子集,对识别有害 sSNV 具有重要意义。我们的结果表明,除了剪接和保守性特征外,新的翻译效率特征也是识别有害 sSNV 的一个信息丰富的特征。虽然功能区域注释和序列特征信息量较弱,但当与其他特征结合使用时,它们可能具有区分有害 sSNV 和良性 sSNV 的能力。数据和源代码可在网站 http://bioinfo.ahu.edu.cn:8080/IDSV 上获得。