School of Environment and Energy, Shenzhen Graduate School, Peking University, Shenzhen, China.
Environmental Biotechnology Laboratory, The University of Hong Kong, Pokfulam road, Hong Kong, China.
Microbiome. 2019 Feb 6;7(1):16. doi: 10.1186/s40168-019-0634-5.
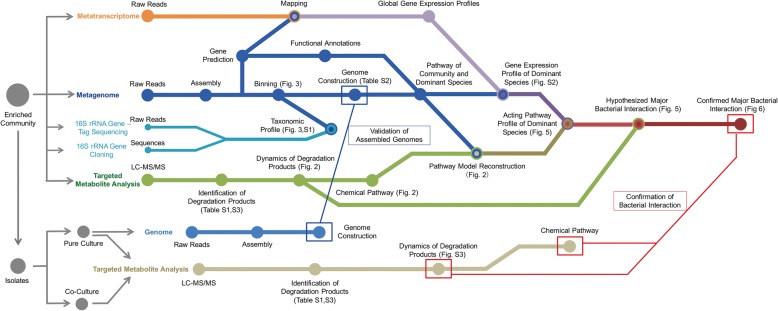
Understanding microbial interactions in engineering bioprocesses is important to enhance and optimize performance outcomes and requires dissection of the multi-layer complexities of microbial communities. However, unraveling microbial interactions as well as substrates involved in complex microbial communities is a challenging task. Here, we demonstrate an integrated approach of metagenomics, metatranscriptomics, and targeted metabolite analysis to identify the substrates involved in interspecies interactions from a potential cross-feeding model community-bisphenol A (BPA)-biodegrading community, aiming to establish an identification method of microbial interactions in engineering or environmental bioprocesses.
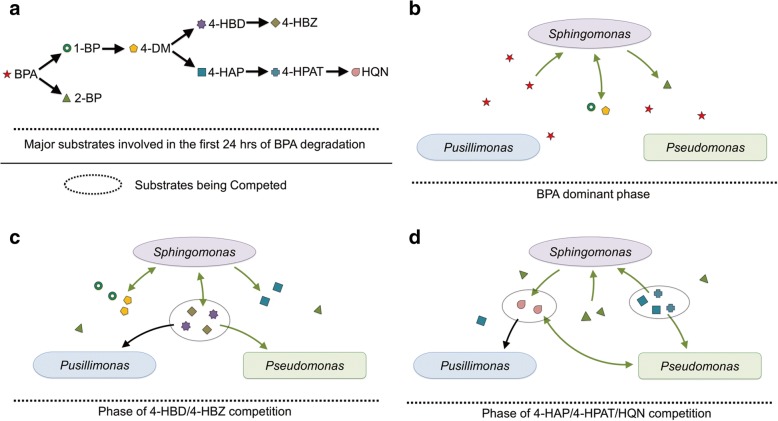
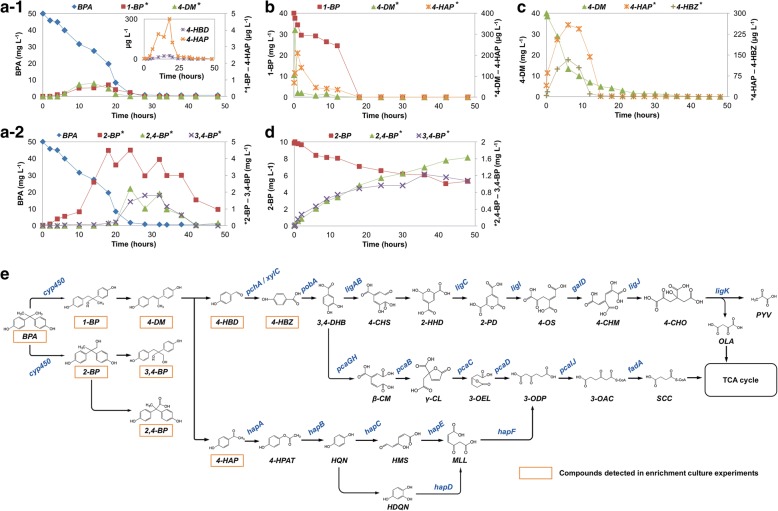
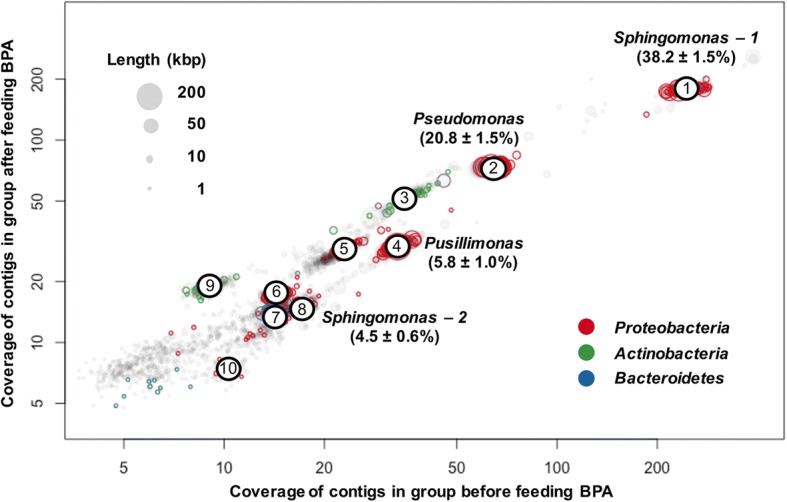
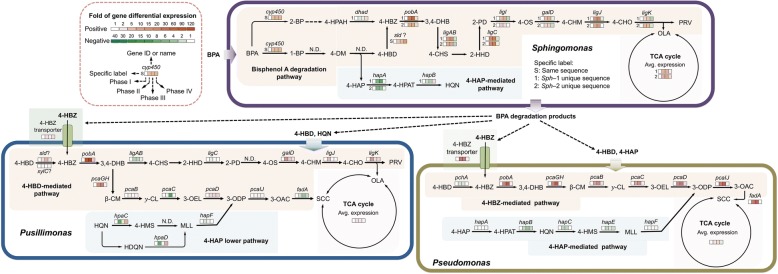
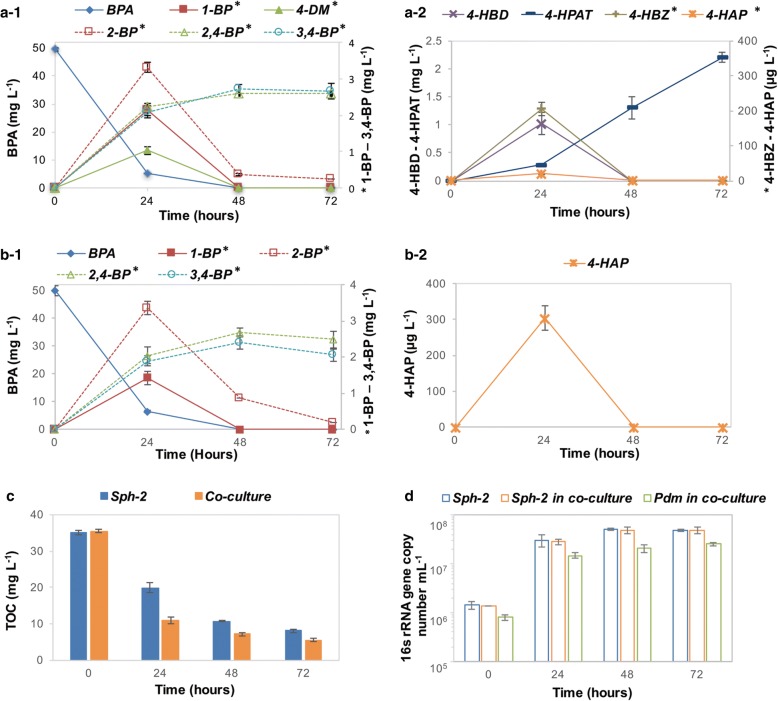
The community-level BPA-metabolic pathway was constructed using integrated metagenomics and targeted metabolite analyses. The dynamics of active functions and metabolism of major community members were identified using metagenomic and metatranscriptomic analyses in concert. Correlating the community BPA biodegradation performance to the individual bacterial activities enabled the discovery of substrates involved in a synergistic interaction of cross-feeding between BPA-degrading Sphingonomas species and intermediate users, Pseudomonas sp. and Pusillimonas sp. This proposed synergistic interaction was confirmed by the co-culture of a Sphingonomas sp. and Pseudomonas sp. isolates, which demonstrated enhanced BPA biodegradation compared to the isolate of Sphingonomas sp. alone.
The three types of integrated meta-omics analyses effectively revealed the metabolic capability at both community-wide and individual bacterial levels. The correlation between these two levels revealed the hidden connection between apparent overall community performance and the contributions of individual community members and their interactions in a BPA-degrading microbial community. In addition, we demonstrated that using integrated multi-omics in conjunction with culture-based confirmation approach is effective to elucidate the microbial interactions affecting the performance outcome. We foresee this approach would contribute the future application and operation of environmental bioprocesses on a knowledge-based control.
了解工程生物工艺中微生物的相互作用对于提高和优化性能结果非常重要,这需要剖析微生物群落的多层复杂性。然而,揭示复杂微生物群落中的微生物相互作用以及涉及的基质是一项具有挑战性的任务。在这里,我们展示了一种综合的宏基因组学、宏转录组学和靶向代谢物分析方法,用于从双酚 A (BPA) 降解群落这一潜在的共营养模型群落中鉴定种间相互作用涉及的基质,旨在建立一种在工程或环境生物工艺中识别微生物相互作用的方法。
使用综合宏基因组学和靶向代谢物分析构建了群落水平的 BPA 代谢途径。使用宏基因组学和宏转录组学分析协同确定了主要群落成员的功能和代谢动力学。将群落 BPA 生物降解性能与单个细菌活性相关联,使我们能够发现共营养种间相互作用中涉及的基质,这种相互作用发生在 BPA 降解的鞘氨醇单胞菌种和中间利用者假单胞菌和 Pusillimonas 种之间。这种协同作用通过 Sphingomonas sp. 和 Pseudomonas sp. 的共培养得到了证实,与 Sphingomonas sp. 单独培养相比,共培养增强了 BPA 的生物降解。
三种类型的综合宏组学分析有效地揭示了在群落范围和单个细菌水平上的代谢能力。这两个层次之间的相关性揭示了明显的总体群落性能与单个群落成员的贡献及其在 BPA 降解微生物群落中的相互作用之间的隐藏联系。此外,我们证明了使用综合多组学结合基于培养的确认方法可以有效地阐明影响性能结果的微生物相互作用。我们预计这种方法将有助于基于知识的控制未来环境生物工艺的应用和运行。