Winter Robin, Montanari Floriane, Noé Frank, Clevert Djork-Arné
Department of Bioinformatics , Bayer AG , Berlin , Germany . Email:
Department of Mathematics and Computer Science , Freie Universität Berlin , Berlin , Germany.
Chem Sci. 2018 Nov 19;10(6):1692-1701. doi: 10.1039/c8sc04175j. eCollection 2019 Feb 14.
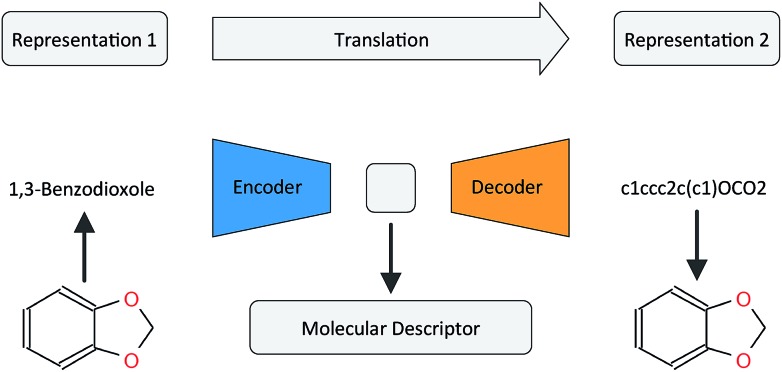
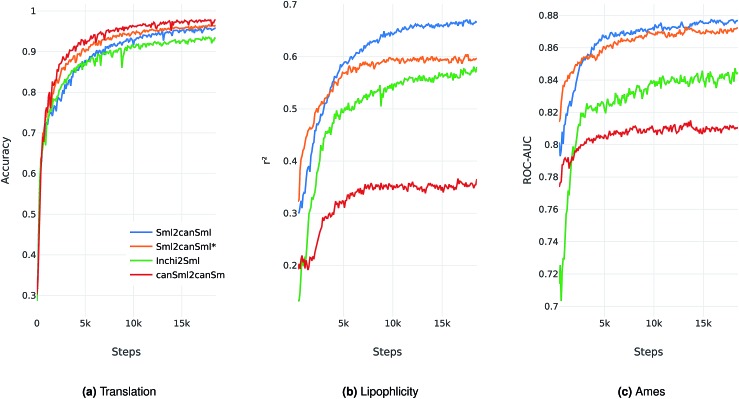
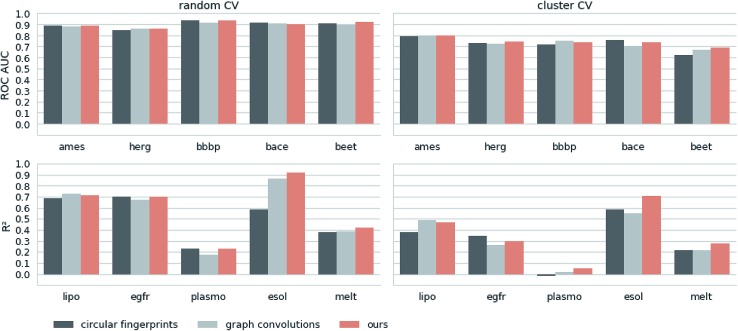
There has been a recent surge of interest in using machine learning across chemical space in order to predict properties of molecules or design molecules and materials with the desired properties. Most of this work relies on defining clever feature representations, in which the chemical graph structure is encoded in a uniform way such that predictions across chemical space can be made. In this work, we propose to exploit the powerful ability of deep neural networks to learn a feature representation from low-level encodings of a huge corpus of chemical structures. Our model borrows ideas from neural machine translation: it translates between two semantically equivalent but syntactically different representations of molecular structures, compressing the meaningful information both representations have in common in a low-dimensional representation vector. Once the model is trained, this representation can be extracted for any new molecule and utilized as a descriptor. In fair benchmarks with respect to various human-engineered molecular fingerprints and graph-convolution models, our method shows competitive performance in modelling quantitative structure-activity relationships in all analysed datasets. Additionally, we show that our descriptor significantly outperforms all baseline molecular fingerprints in two ligand-based virtual screening tasks. Overall, our descriptors show the most consistent performances in all experiments. The continuity of the descriptor space and the existence of the decoder that permits deducing a chemical structure from an embedding vector allow for exploration of the space and open up new opportunities for compound optimization and idea generation.
最近,利用机器学习在化学空间中预测分子性质或设计具有所需性质的分子和材料的兴趣激增。这项工作大多依赖于定义巧妙的特征表示,其中化学图结构以统一的方式进行编码,以便能够对化学空间进行预测。在这项工作中,我们建议利用深度神经网络的强大能力,从大量化学结构的低级编码中学习特征表示。我们的模型借鉴了神经机器翻译的思想:它在分子结构的两种语义等效但句法不同的表示之间进行转换,将两种表示共有的有意义信息压缩到一个低维表示向量中。一旦模型经过训练,就可以为任何新分子提取这种表示并用作描述符。在针对各种人工设计的分子指纹和图卷积模型的公平基准测试中,我们的方法在所有分析数据集中的定量构效关系建模方面表现出具有竞争力的性能。此外,我们表明,在两项基于配体的虚拟筛选任务中,我们的描述符明显优于所有基线分子指纹。总体而言,我们的描述符在所有实验中表现出最一致的性能。描述符空间的连续性以及允许从嵌入向量推导出化学结构的解码器的存在,为探索该空间提供了可能,并为化合物优化和新想法的产生开辟了新机会。