Department of Microbiology, The University of Hong Kong, Hong Kong, China.
Department of Microbiology, The University of Hong Kong, Hong Kong, China; State Key Laboratory of Emerging Infectious Diseases, The University of Hong Kong, Hong Kong, China; Carol Yu Centre for Infection, The University of Hong Kong, Hong Kong, China; Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, Zhejiang University, Hangzhou 310006, China.
Infect Genet Evol. 2019 Jul;71:21-30. doi: 10.1016/j.meegid.2019.03.001. Epub 2019 Mar 4.
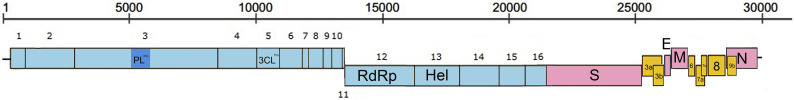
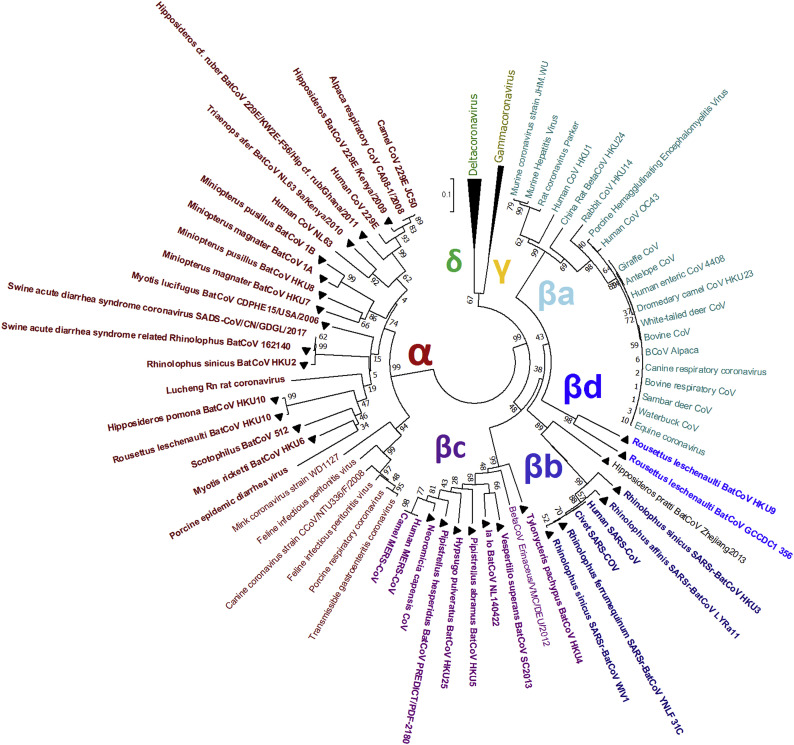
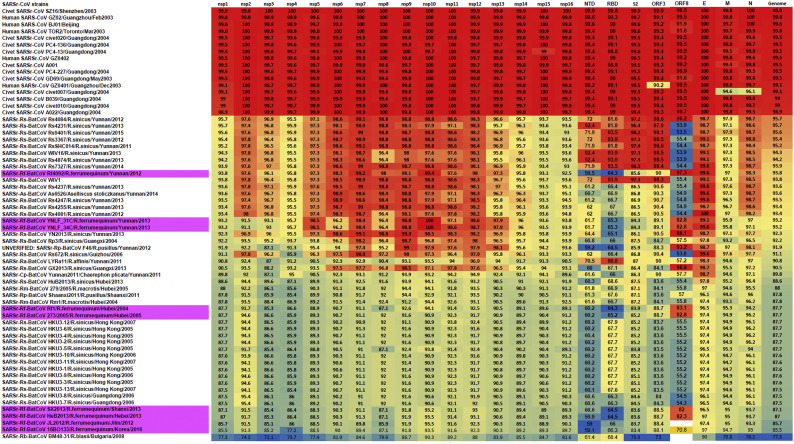
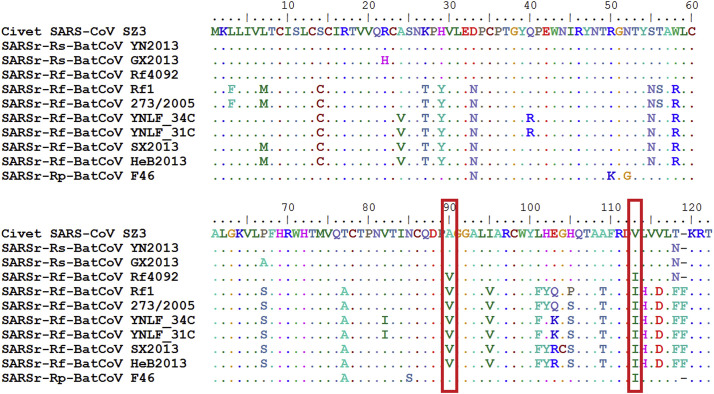
Shortly after its emergence in southern China in 2002/2003, Severe Acute Respiratory Syndrome coronavirus (SARS-CoV) was confirmed to be the cause of SARS. Subsequently, SARS-related CoVs (SARSr-CoVs) were found in palm civets from live animal markets in Guangdong and in various horseshoe bat species, which were believed to be the ultimate reservoir of SARSr-CoV. Till November 2018, 339 SARSr-CoV genomes have been sequenced, including 274 from human, 18 from civets and 47 from bats [mostly from Chinese horseshoe bats (Rhinolophus sinicus), n = 30; and greater horseshoe bats (Rhinolophus ferrumequinum), n = 9]. The human SARS-CoVs and civet SARSr-CoVs were collected in 2003/2004, while bat SARSr-CoVs were continuously isolated in the past 13 years even after the cessation of the SARS epidemic. SARSr-CoVs belong to the subgenus Sarbecovirus (previously lineage B) of genus Betacoronavirus and occupy a unique phylogenetic position. Overall, it is observed that the SARSr-CoV genomes from bats in Yunnan province of China possess the highest nucleotide identity to those from civets. It is evident from both multiple alignment and phylogenetic analyses that some genes of a particular SARSr-CoV from bats may possess higher while other genes possess much lower nucleotide identity to the corresponding genes of SARSr-CoV from human/civets, resulting in the shift of phylogenetic position in different phylogenetic trees. Our current model on the origin of SARS is that the human SARS-CoV that caused the epidemic in 2002/2003 was probably a result of multiple recombination events from a number of SARSr-CoV ancestors in different horseshoe bat species.
2002/2003 年在中国南方出现后不久,严重急性呼吸综合征冠状病毒(SARS-CoV)被确认为 SARS 的病因。随后,在广东的活体动物市场中的果子狸和各种马蹄蝠中发现了与 SARS 相关的冠状病毒(SARSr-CoV),这些动物被认为是 SARSr-CoV 的最终宿主。截至 2018 年 11 月,已对 339 个 SARSr-CoV 基因组进行了测序,其中 274 个来自人类,18 个来自果子狸,47 个来自蝙蝠[主要来自中华菊头蝠(Rhinolophus sinicus),n=30;和大足鼠耳蝠(Rhinolophus ferrumequinum),n=9]。人类 SARS-CoV 和果子狸 SARSr-CoV 是在 2003/2004 年收集的,而蝙蝠 SARSr-CoV 则在过去 13 年中不断被分离出来,即使在 SARS 疫情停止之后也是如此。SARSr-CoV 属于贝塔冠状病毒属的Sarbecovirus 亚属(以前称为谱系 B),占据独特的系统发育位置。总体而言,中国云南省蝙蝠的 SARSr-CoV 基因组与果子狸的 SARSr-CoV 基因组具有最高的核苷酸同一性。从多重比对和系统发育分析可以明显看出,来自蝙蝠的特定 SARSr-CoV 的某些基因可能具有较高的核苷酸同一性,而其他基因则与来自人类/果子狸的 SARSr-CoV 的相应基因具有较低的核苷酸同一性,从而导致不同系统发育树中的系统发育位置发生变化。我们目前关于 SARS 起源的模型是,2002/2003 年引起流行的人类 SARS-CoV 可能是来自不同马蹄蝠物种的多种 SARSr-CoV 祖先的多次重组事件的结果。