Bo Hao, Cao Ke, Tang Ruiling, Zhang Han, Gong Zhaojian, Liu Zhizhong, Liu Jianye, Li Jingjing, Fan Liqing
Institute of Reproductive and Stem Cell Engineering, School of Basic Medical Science, Central South University, Changsha, China.
Department of Oncology, Third Xiangya Hospital, Central South University, Changsha, Hunan, China.
J Cancer. 2019 Jan 29;10(4):893-902. doi: 10.7150/jca.27491. eCollection 2019.
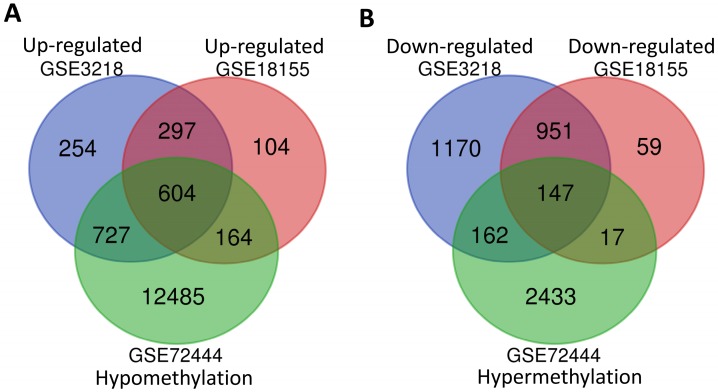
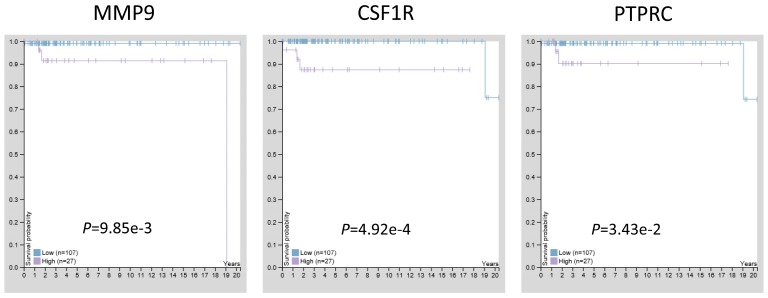
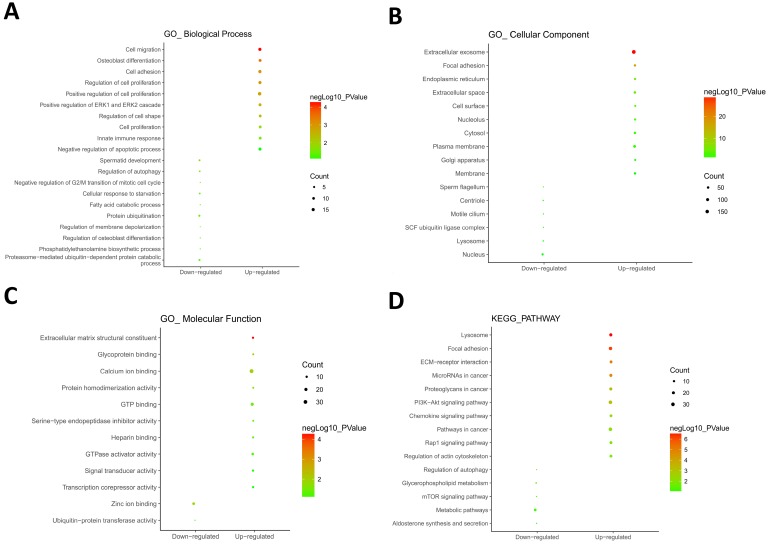
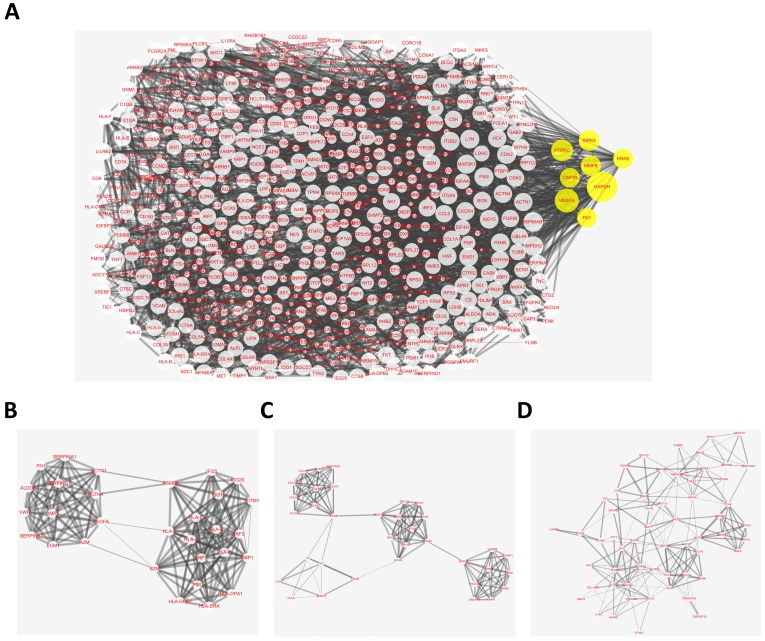
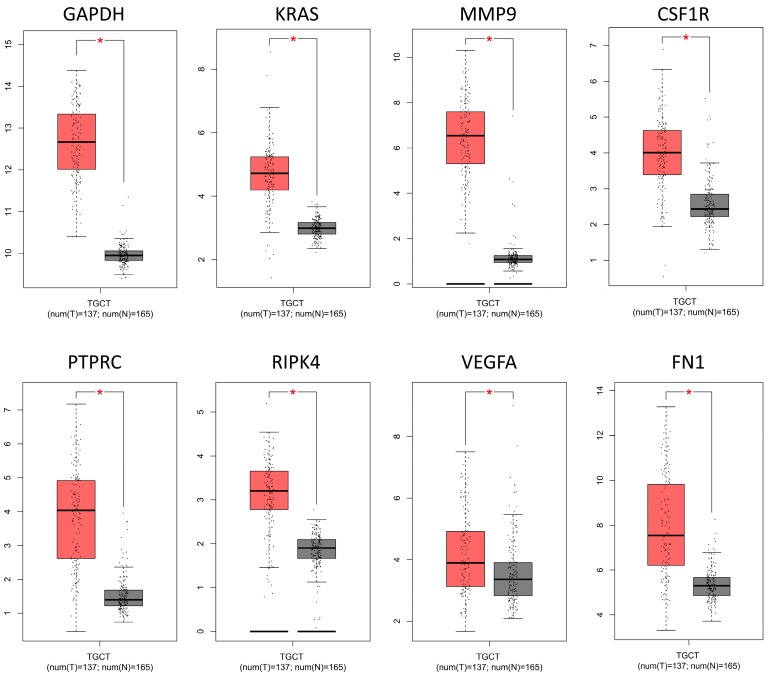
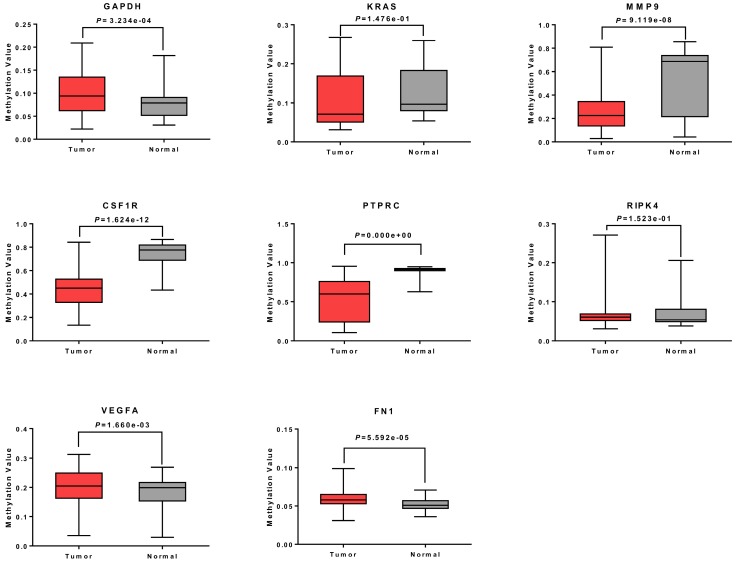
: Testicular germ cell tumors (TGCT) is the most common testicular malignancy threaten young male reproductive health. This study aimed to identify aberrantly methylated-differentially expressed genes and pathways in TGCT by comprehensive bioinformatics analysis. : Data of gene expression microarrays (GSE3218, GSE18155) and gene methylation microarrays (GSE72444) were collected from GEO database. Integrated analysis acquired aberrantly methylated-genes. Functional and pathway enrichment analysis were performed using DAVID database. Protein-protein interaction (PPI) network was constructed by STRING and App Mcode was used for module analysis. GEPIA platform and DiseaseMeth database were used for confirming the expression and methylation levels of hub genes. Finally, Human Protein Atlas database was performed to evaluate the prognostic significance. : Totally 604 hypomethylation-high expression and 147 hypermethylation-low genes were identified. The high expressed genes were enriched in biological processes of cell proliferation and migration. The top 8 hub genes of PPI network were GAPDH, VEGFA, PTPRC, RIPK4, MMP9, CSF1R, KRAS and FN1. After validation in GEPIA platform, all hub genes were elevated in TGCT tissues. Only MMP9, CSF1R and PTPRC showed hypomethylation-high expression status, which predicted the poor outcome of TGCT patients. : Our study indicated possible aberrantly methylated-differentially expressed genes and pathways in TGCT by bioinformatics analysis, which may provide novel insights for unraveling pathogenesis of TGCT.
睾丸生殖细胞肿瘤(TGCT)是威胁年轻男性生殖健康的最常见睾丸恶性肿瘤。本研究旨在通过综合生物信息学分析鉴定TGCT中异常甲基化差异表达的基因和通路。
从基因表达综合数据库(GEO)收集基因表达微阵列(GSE3218、GSE18155)和基因甲基化微阵列(GSE72444)的数据。综合分析获得异常甲基化基因。使用DAVID数据库进行功能和通路富集分析。通过STRING构建蛋白质-蛋白质相互作用(PPI)网络,并使用App Mcode进行模块分析。利用GEPIA平台和疾病甲基化数据库确认枢纽基因的表达和甲基化水平。最后,通过人类蛋白质图谱数据库评估预后意义。
共鉴定出604个低甲基化高表达基因和147个高甲基化低表达基因。高表达基因富集于细胞增殖和迁移的生物学过程。PPI网络的前8个枢纽基因是甘油醛-3-磷酸脱氢酶(GAPDH)、血管内皮生长因子A(VEGFA)、蛋白酪氨酸磷酸酶受体C(PTPRC)、受体相互作用丝氨酸/苏氨酸蛋白激酶4(RIPK4)、基质金属蛋白酶9(MMP9)、集落刺激因子1受体(CSF1R)、 Kirsten大鼠肉瘤病毒癌基因同源物(KRAS)和纤连蛋白1(FN1)。在GEPIA平台验证后,所有枢纽基因在TGCT组织中均升高。只有MMP9、CSF1R和PTPRC表现出低甲基化高表达状态,这预示着TGCT患者预后不良。
我们的研究通过生物信息学分析表明TGCT中可能存在异常甲基化差异表达的基因和通路,这可能为揭示TGCT的发病机制提供新的见解。