Exobiology Branch, NASA Ames Research Center, Moffett Field, CA, USA.
Bioinformatics. 2019 Oct 15;35(20):4162-4164. doi: 10.1093/bioinformatics/btz188.
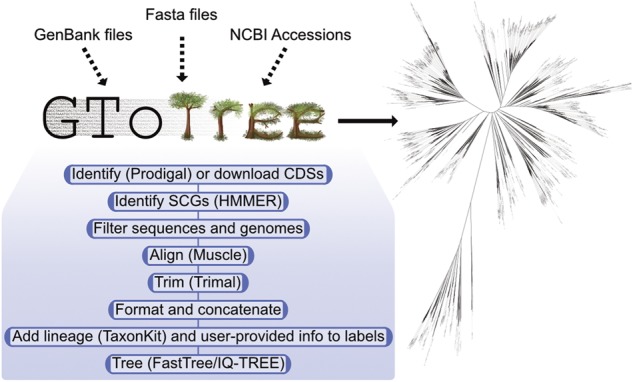
Genome-level evolutionary inference (i.e. phylogenomics) is becoming an increasingly essential step in many biologists' work. Accordingly, there are several tools available for the major steps in a phylogenomics workflow. But for the biologist whose main focus is not bioinformatics, much of the computational work required-such as accessing genomic data on large scales, integrating genomes from different file formats, performing required filtering, stitching different tools together etc.-can be prohibitive. Here I introduce GToTree, a command-line tool that can take any combination of fasta files, GenBank files and/or NCBI assembly accessions as input and outputs an alignment file, estimates of genome completeness and redundancy, and a phylogenomic tree based on a specified single-copy gene (SCG) set. Although GToTree can work with any custom hidden Markov Models (HMMs), also included are 13 newly generated SCG-set HMMs for different lineages and levels of resolution, built based on searches of ∼12 000 bacterial and archaeal high-quality genomes. GToTree aims to give more researchers the capability to make phylogenomic trees.
GToTree is open-source and freely available for download from: github.com/AstrobioMike/GToTree. It is implemented primarily in bash with helper scripts written in python.
Supplementary data are available at Bioinformatics online.
基因组水平的进化推断(即系统基因组学)在许多生物学家的工作中变得越来越重要。因此,有几个工具可用于系统基因组学工作流程的主要步骤。但是,对于主要关注点不是生物信息学的生物学家来说,许多计算工作(例如大规模访问基因组数据、整合来自不同文件格式的基因组、执行所需的过滤、将不同的工具拼接在一起等)可能是不可行的。在这里,我介绍了 GToTree,这是一个命令行工具,可以接受任何组合的 fasta 文件、GenBank 文件和/或 NCBI 组装访问号作为输入,并输出一个对齐文件、基因组完整性和冗余度的估计值,以及基于指定的单拷贝基因 (SCG) 集的系统发育树。虽然 GToTree 可以与任何自定义隐马尔可夫模型 (HMM) 一起使用,但也包括 13 个新生成的 SCG 集 HMM,用于不同的谱系和分辨率级别,这些 HMM 是基于对大约 12000 个细菌和古细菌高质量基因组的搜索构建的。GToTree 的目标是让更多的研究人员能够构建系统发育树。
GToTree 是开源的,可从 github.com/AstrobioMike/GToTree 免费下载。它主要用 bash 编写,并使用 python 编写辅助脚本实现。
补充数据可在 Bioinformatics 在线获取。