Department of Pathogen Biology, College of Basic Medical Sciences, Jilin University, Changchun, China.
Department of Respiratory and Sleep Medicine, John Hunter Hospital, Newcastle, Australia.
Int J Med Sci. 2019 Mar 9;16(3):477-485. doi: 10.7150/ijms.29433. eCollection 2019.
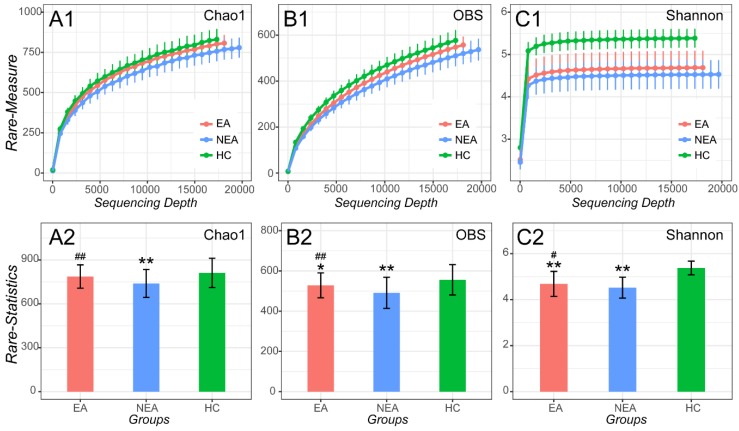
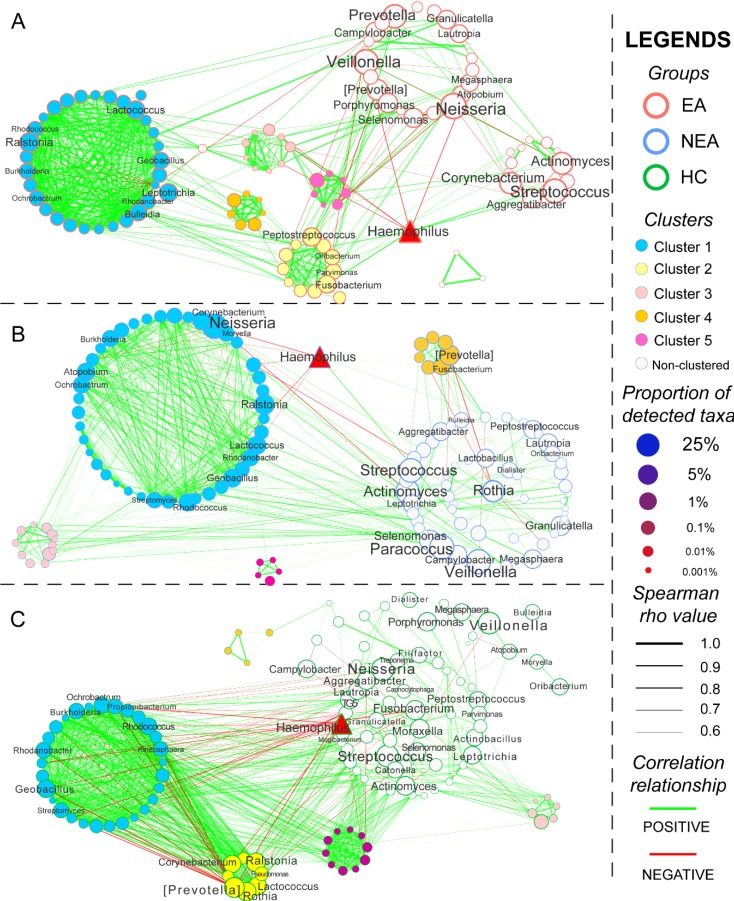
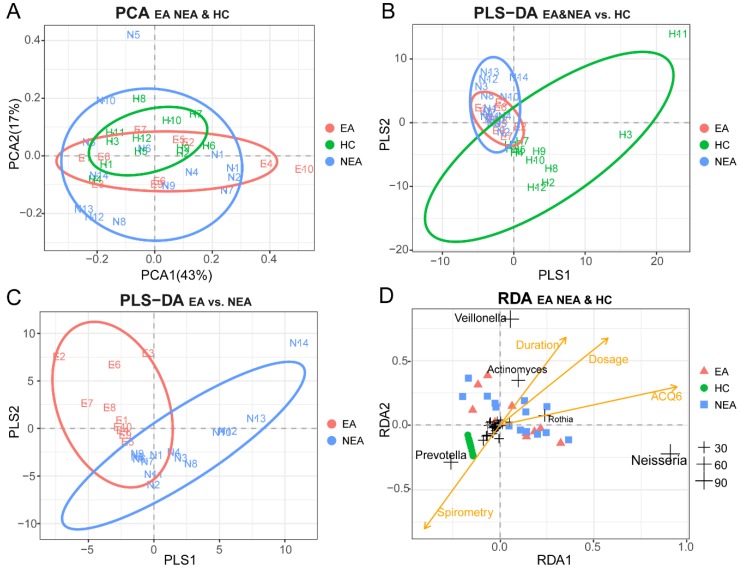
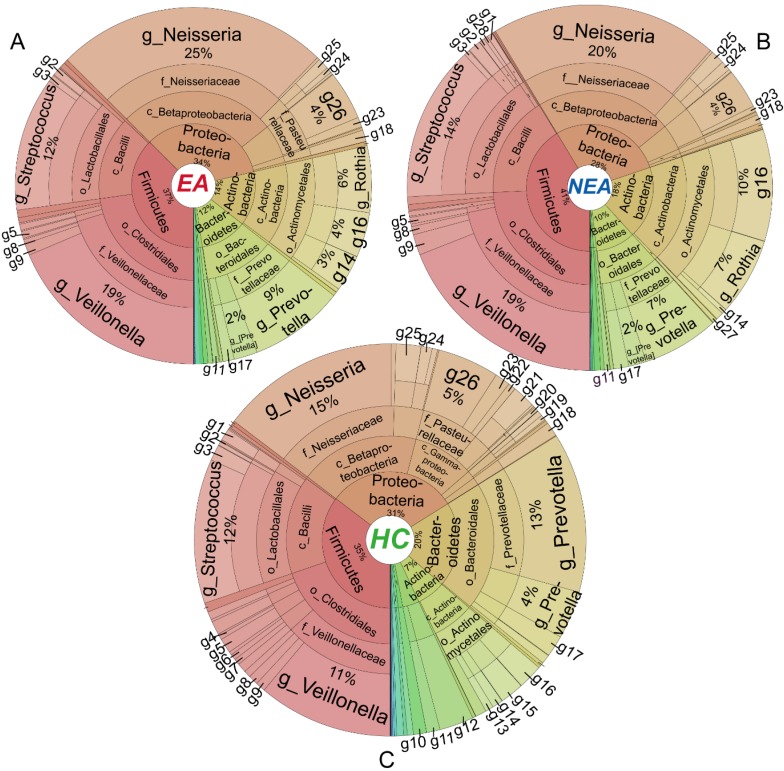
Asthma is a common respiratory disease with a high prevalence and morbidity that can seriously affect quality of life. Microbial colonization of the airway may participate in the pathogenesis of asthma, however the mechanisms involved have not been established. In the present study, we aimed to determine the composition of the microbiota in different asthmatic phenotypes from Northeast China. 24 mild-to-moderate asthmatics (10 eosinophilic asthma and 14 non-eosinophilic asthma) and 12 healthy volunteers participated in this cross-sectional study. DNA was extracted from their induced sputum and amplified for 16s rRNA gene sequencing on Illumina Miseq platform. Bioinformatic analysis on the microbiome was performed. Alpha-diversity analysis showed that the asthmatics had a decreased richness, evenness and diversity. Non-eosinophilic asthmatics showed a decreased richness, evenness and diversity compared with eosinophilic patients. A different taxonomy of 1 phylum and 6 genera taxa between the phenotypes was identified. Compared with heathy controls, asthmatics existed a larger taxonomic difference (<0.05 for both EA and NEA vs. HC). 5 genera as the dominance in the microbial co-occurrence network correlated with the spirometry and disease progression of asthma. The function of microbiota genes was predicted to be related with infectious, immune and metabolic diseases. The diversity and composition of the airway microbiome was associated with the pathogenesis of asthma in different phenotypes. The diverse composition has been identified in the present study.
哮喘是一种常见的呼吸道疾病,患病率和发病率高,可严重影响生活质量。气道微生物定植可能参与哮喘的发病机制,但相关机制尚未确定。本研究旨在确定来自中国东北地区不同哮喘表型的微生物群组成。 24 名轻度至中度哮喘患者(10 名嗜酸性粒细胞性哮喘和 14 名非嗜酸性粒细胞性哮喘)和 12 名健康志愿者参与了这项横断面研究。从他们的诱导痰中提取 DNA,并在 Illumina Miseq 平台上对 16s rRNA 基因进行扩增测序。对微生物组进行生物信息学分析。 多样性分析表明,哮喘患者的丰富度、均匀度和多样性降低。与嗜酸性粒细胞性患者相比,非嗜酸性粒细胞性哮喘患者的丰富度、均匀度和多样性降低。表型之间确定了 1 个门和 6 个属的分类差异。与健康对照组相比,哮喘患者的分类差异更大(EA 和 NEA 均<0.05 与 HC 相比)。 5 个属作为微生物共同发生网络的优势与哮喘的肺功能和疾病进展相关。预测微生物群基因的功能与传染病、免疫和代谢疾病有关。 气道微生物群的多样性和组成与不同表型的哮喘发病机制有关。本研究中已确定了不同的组成。