Department of Theoretical Biophysics , Max Planck Institute of Biophysics , Max-von-Laue-Straße 3 , 60438 Frankfurt am Main , Germany.
Max Planck Computing and Data Facility , Gießenbachstr. 2 , 85748 Garching , Germany.
J Chem Theory Comput. 2019 May 14;15(5):3390-3401. doi: 10.1021/acs.jctc.8b01231. Epub 2019 Apr 17.
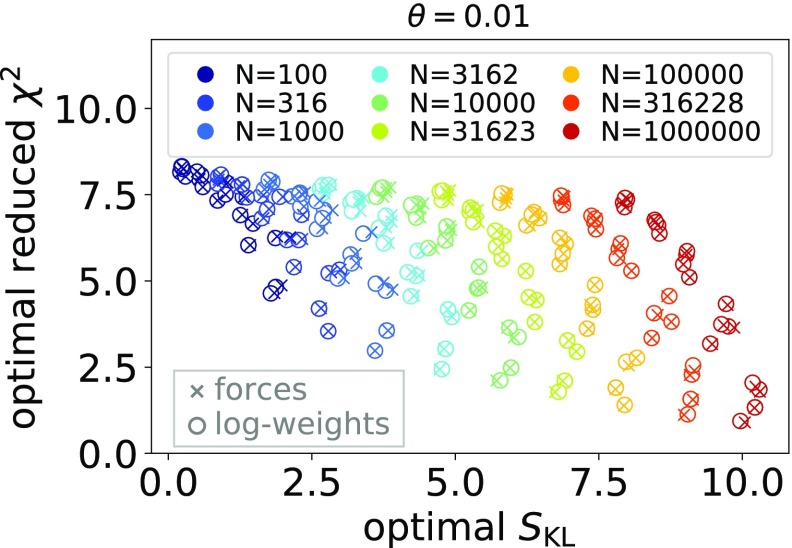
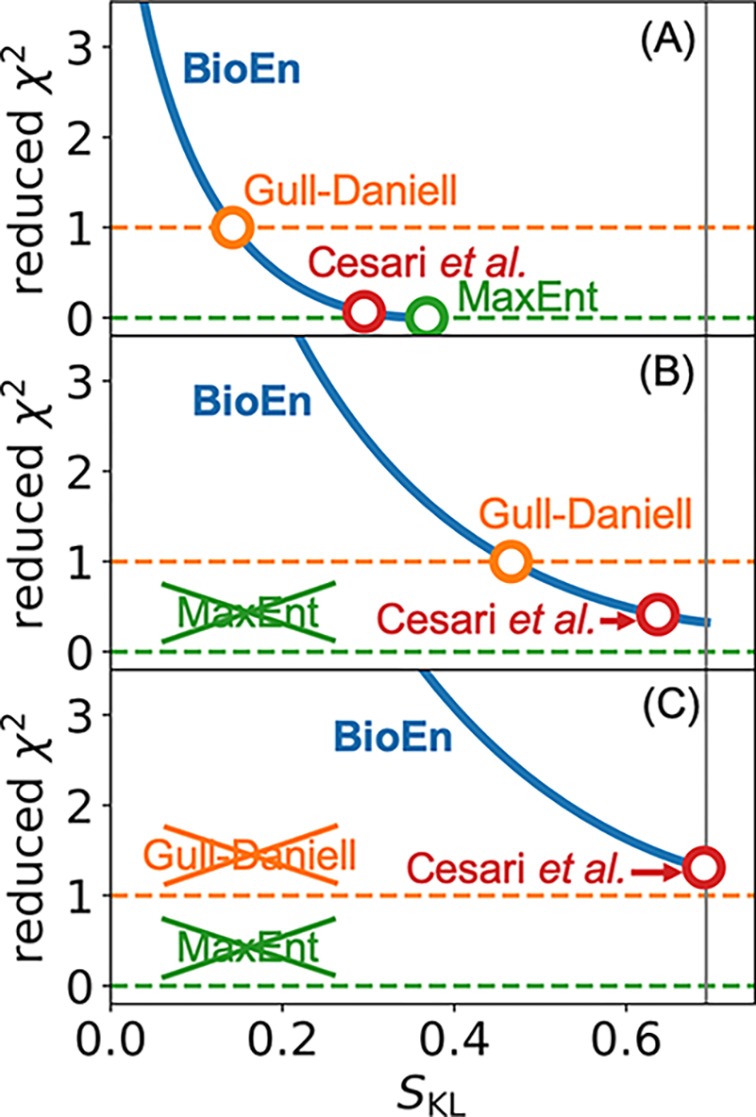
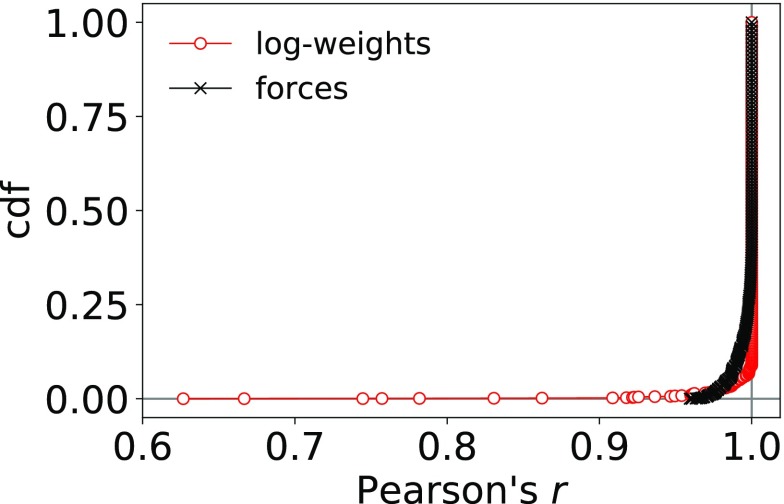
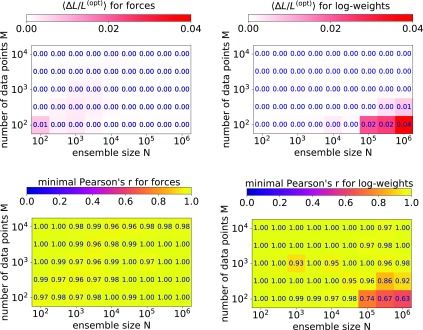
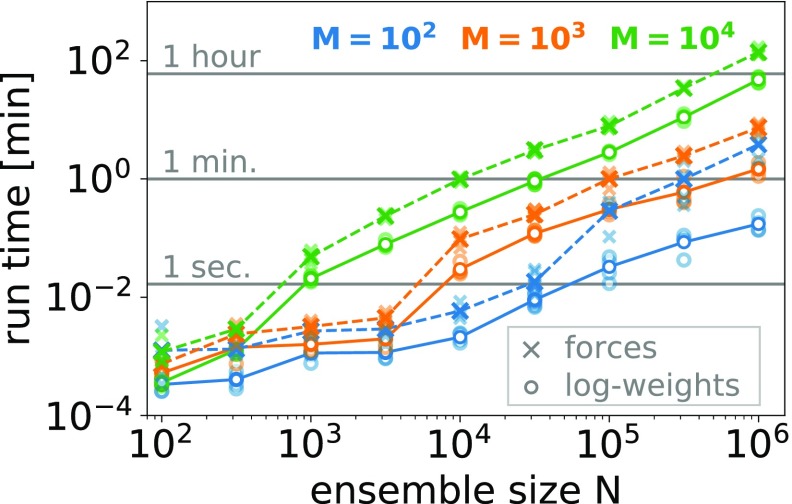
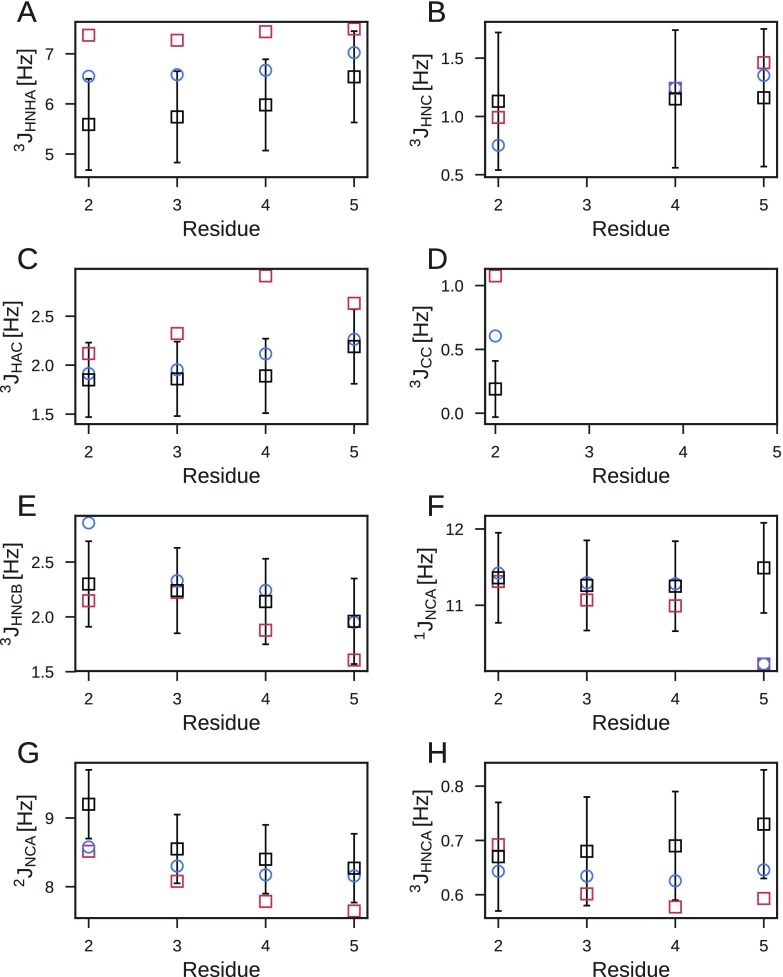
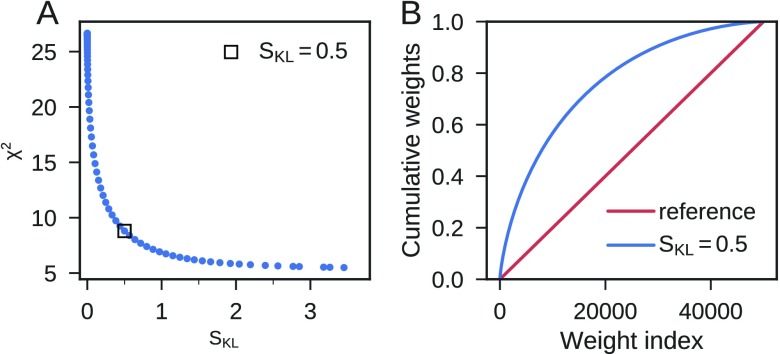
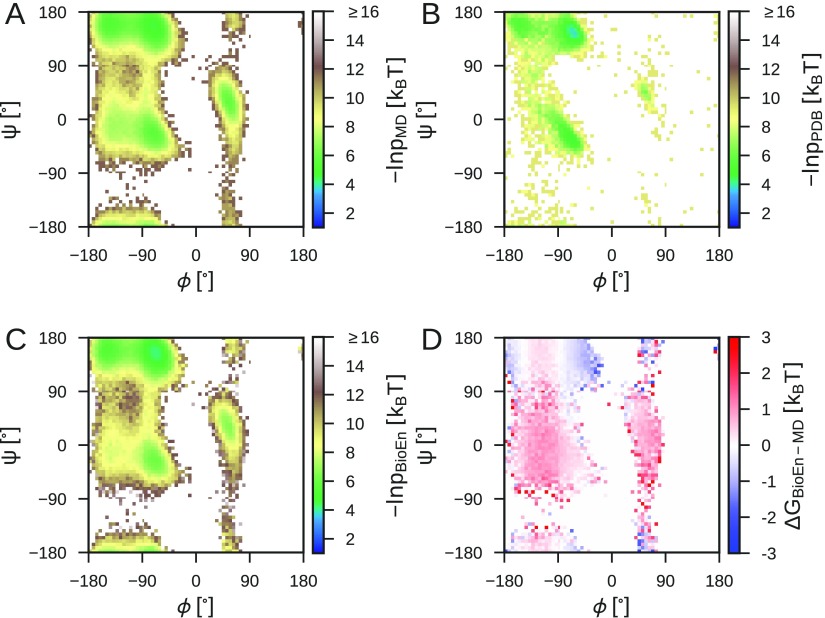
Ensemble refinement produces structural ensembles of flexible and dynamic biomolecules by integrating experimental data and molecular simulations. Here we present two efficient numerical methods to solve the computationally challenging maximum-entropy problem arising from a Bayesian formulation of ensemble refinement. Recasting the resulting constrained weight optimization problem into an unconstrained form enables the use of gradient-based algorithms. In two complementary formulations that differ in their dimensionality, we optimize either the log-weights directly or the generalized forces appearing in the explicit analytical form of the solution. We first demonstrate the robustness, accuracy, and efficiency of the two methods using synthetic data. We then use NMR J-couplings to reweight an all-atom molecular dynamics simulation ensemble of the disordered peptide Ala-5 simulated with the AMBER99SB*-ildn-q force field. After reweighting, we find a consistent increase in the population of the polyproline-II conformations and a decrease of α-helical-like conformations. Ensemble refinement makes it possible to infer detailed structural models for biomolecules exhibiting significant dynamics, such as intrinsically disordered proteins, by combining input from experiment and simulation in a balanced manner.
通过整合实验数据和分子模拟,集合精修可以生成灵活和动态生物分子的结构集合。在这里,我们提出了两种有效的数值方法来解决集合精修的贝叶斯公式中出现的具有挑战性的最大熵问题。将得到的受约束的权重优化问题转换为无约束形式,使得可以使用基于梯度的算法。在两种互补的形式中,它们在维度上有所不同,我们可以直接优化对数权重,或者优化出现在解决方案的显式解析形式中的广义力。我们首先使用合成数据来演示这两种方法的鲁棒性、准确性和效率。然后,我们使用 NMR J 耦合来重新加权无序肽 Ala-5 的全原子分子动力学模拟集合,该肽模拟使用 AMBER99SB*-ildn-q 力场。重新加权后,我们发现聚脯氨酸-II 构象的数量持续增加,α-螺旋样构象的数量减少。通过以平衡的方式将实验和模拟的输入结合起来,集合精修使得对表现出显著动力学的生物分子(如天然无序蛋白)进行详细的结构模型推断成为可能。