Jensen Jan H
Department of Chemistry , University of Copenhagen , Copenhagen , Denmark . Email:
Chem Sci. 2019 Feb 11;10(12):3567-3572. doi: 10.1039/c8sc05372c. eCollection 2019 Mar 28.
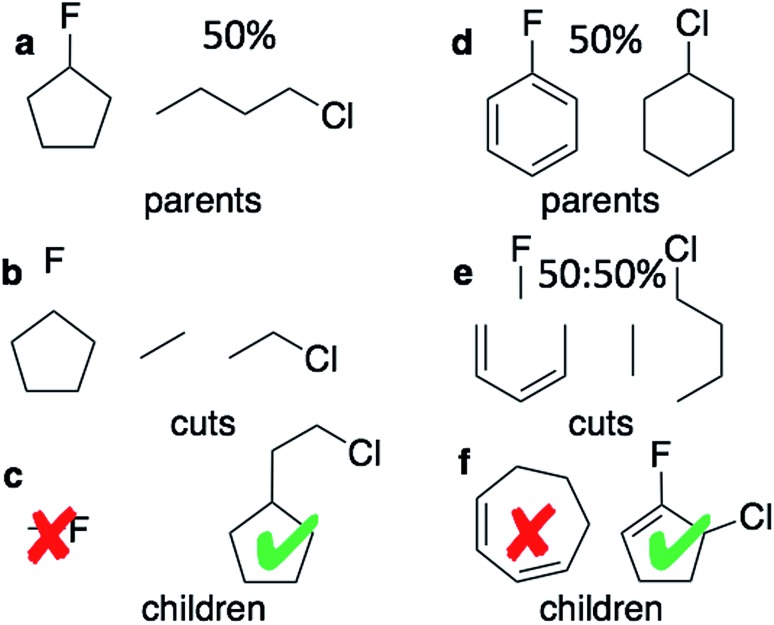
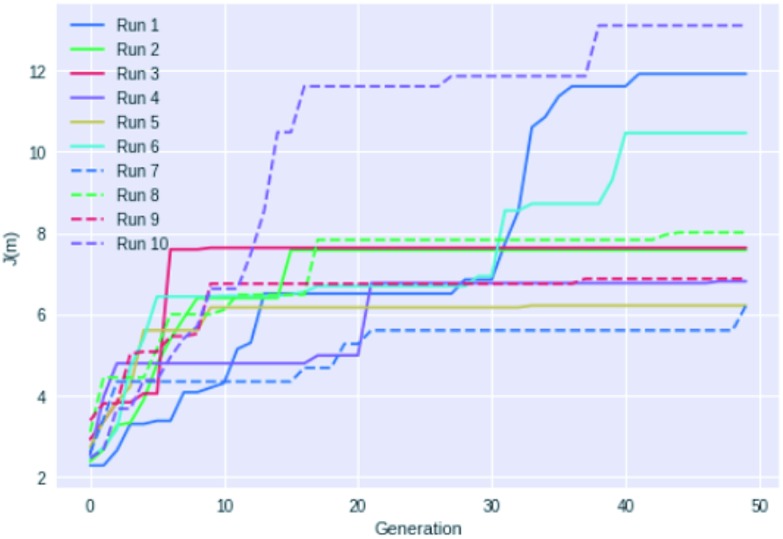
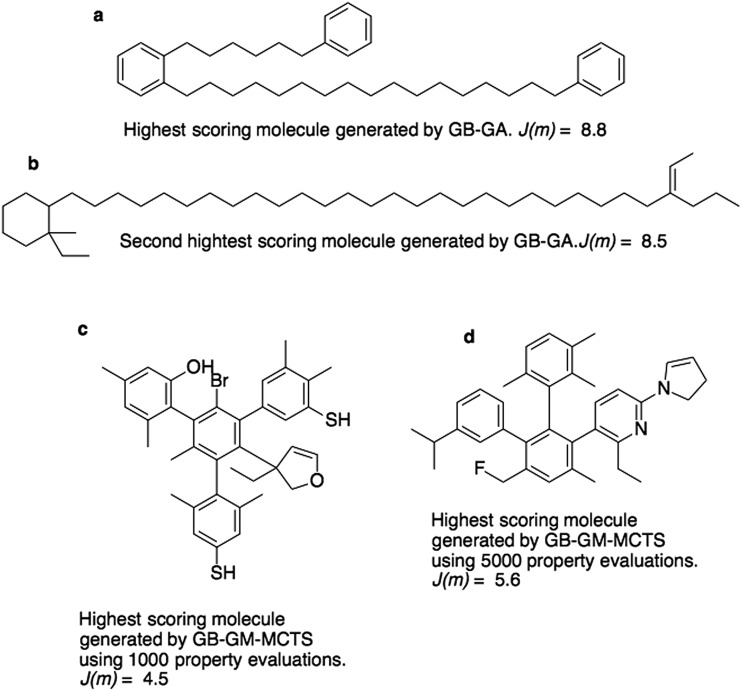
This paper presents a comparison of a graph-based genetic algorithm (GB-GA) and machine learning (ML) results for the optimization of log values with a constraint for synthetic accessibility and shows that the GA is as good as or better than the ML approaches for this particular property. The molecules found by the GB-GA bear little resemblance to the molecules used to construct the initial mating pool, indicating that the GB-GA approach can traverse a relatively large distance in chemical space using relatively few (50) generations. The paper also introduces a new non-ML graph-based generative model (GB-GM) that can be parameterized using very small data sets and combined with a Monte Carlo tree search (MCTS) algorithm. The results are comparable to previously published results (, 2017, , 972-976) using a recurrent neural network (RNN) generative model, and the GB-GM-based method is several orders of magnitude faster. The MCTS results seem more dependent on the composition of the training set than the GA approach for this particular property. Our results suggest that the performance of new ML-based generative models should be compared to that of more traditional, and often simpler, approaches such a GA.
本文对基于图的遗传算法(GB-GA)和机器学习(ML)在优化log 值时的结果进行了比较,同时考虑了合成可及性的约束条件,并表明对于这一特定性质,遗传算法与机器学习方法表现相当甚至更优。GB-GA找到的分子与用于构建初始交配池的分子几乎没有相似之处,这表明GB-GA方法能够在相对较少的(50)代内跨越化学空间中相对较大的距离。本文还介绍了一种新的基于图的非机器学习生成模型(GB-GM),该模型可以使用非常小的数据集进行参数化,并与蒙特卡罗树搜索(MCTS)算法相结合。其结果与之前使用递归神经网络(RNN)生成模型发表的结果(,2017,,972 - 976)相当,并且基于GB-GM的方法要快几个数量级。对于这一特定性质,MCTS的结果似乎比GA方法更依赖于训练集的组成。我们的结果表明,新的基于机器学习的生成模型的性能应该与更传统且通常更简单的方法(如GA)进行比较。