Department of Chemical and Systems Biology, Stanford University School of Medicine, Stanford, CA 94305, USA.
Department of Chemical and Systems Biology, Stanford University School of Medicine, Stanford, CA 94305, USA; Department of Developmental Biology, Stanford University School of Medicine, Stanford, CA 94305, USA.
Cell Syst. 2019 May 22;8(5):363-379.e3. doi: 10.1016/j.cels.2019.04.002. Epub 2019 May 1.
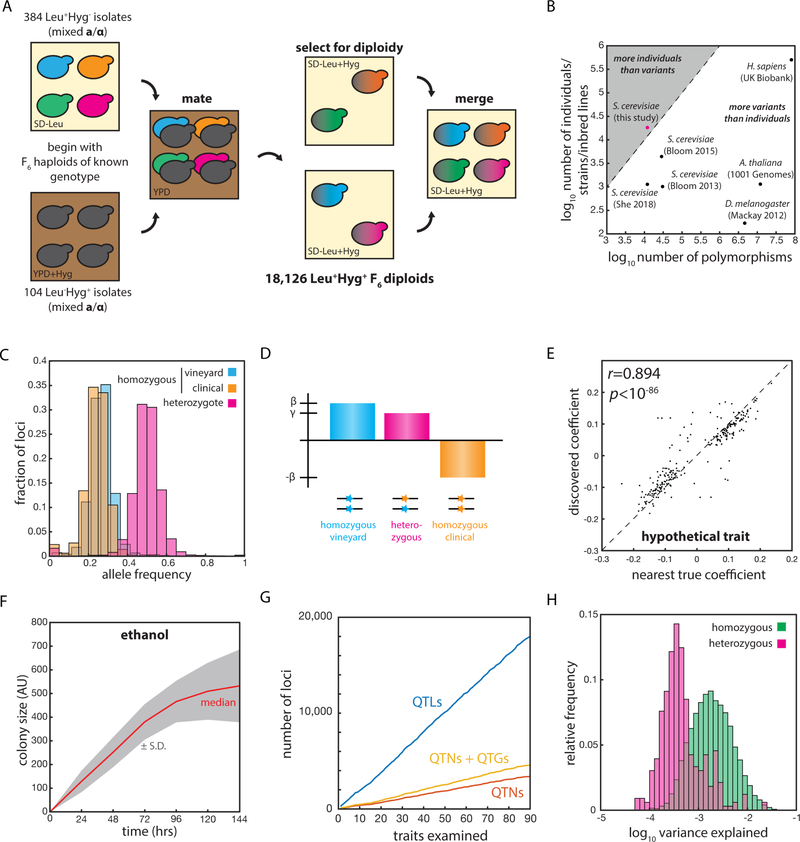
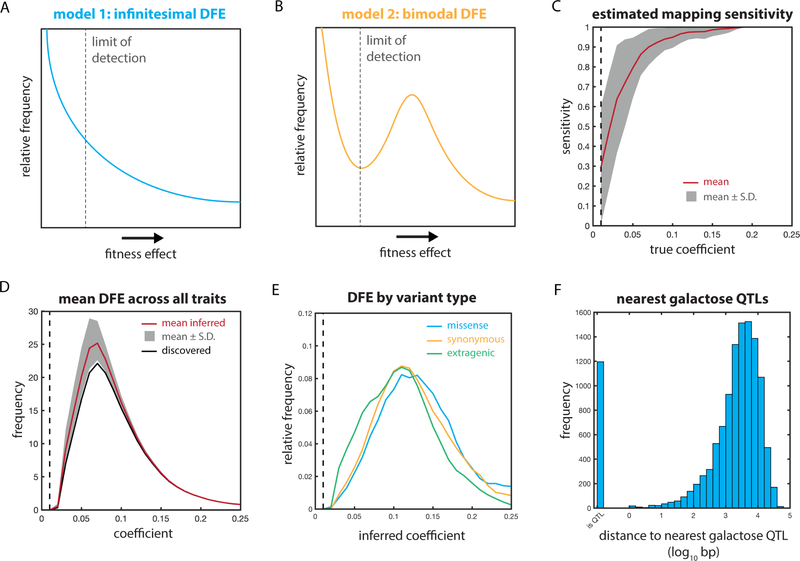
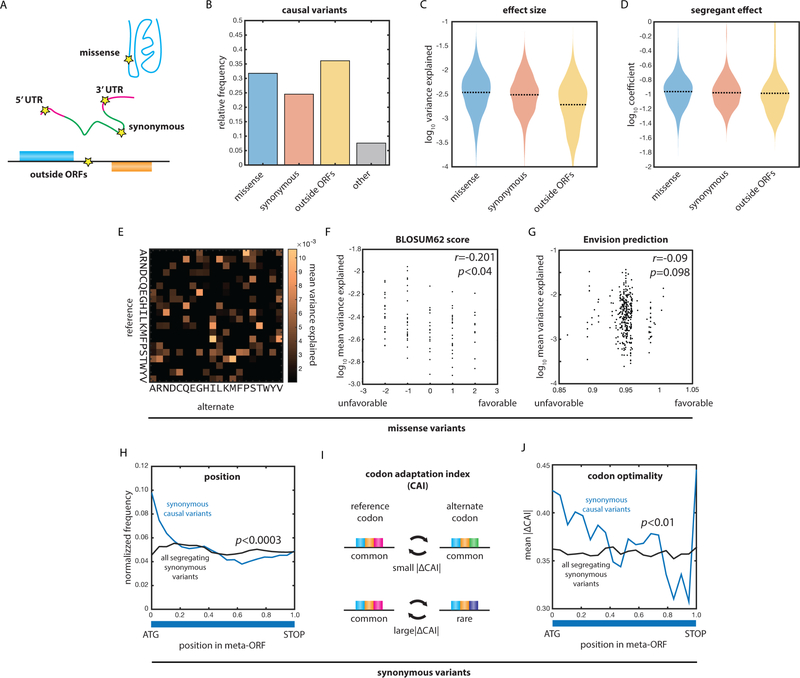
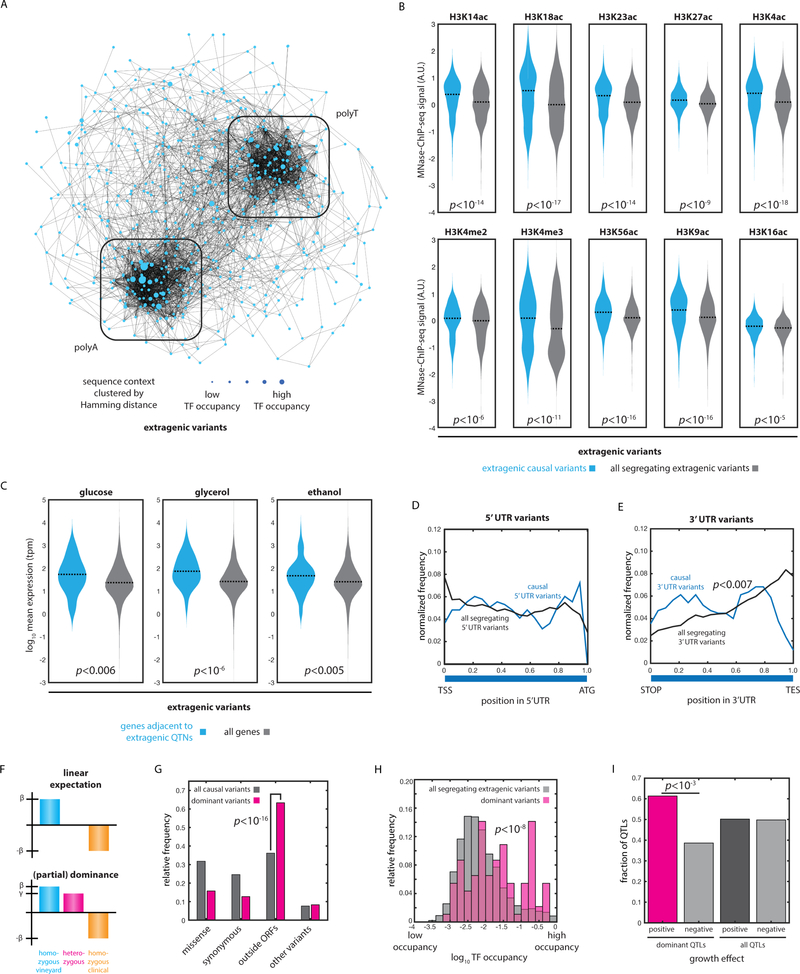
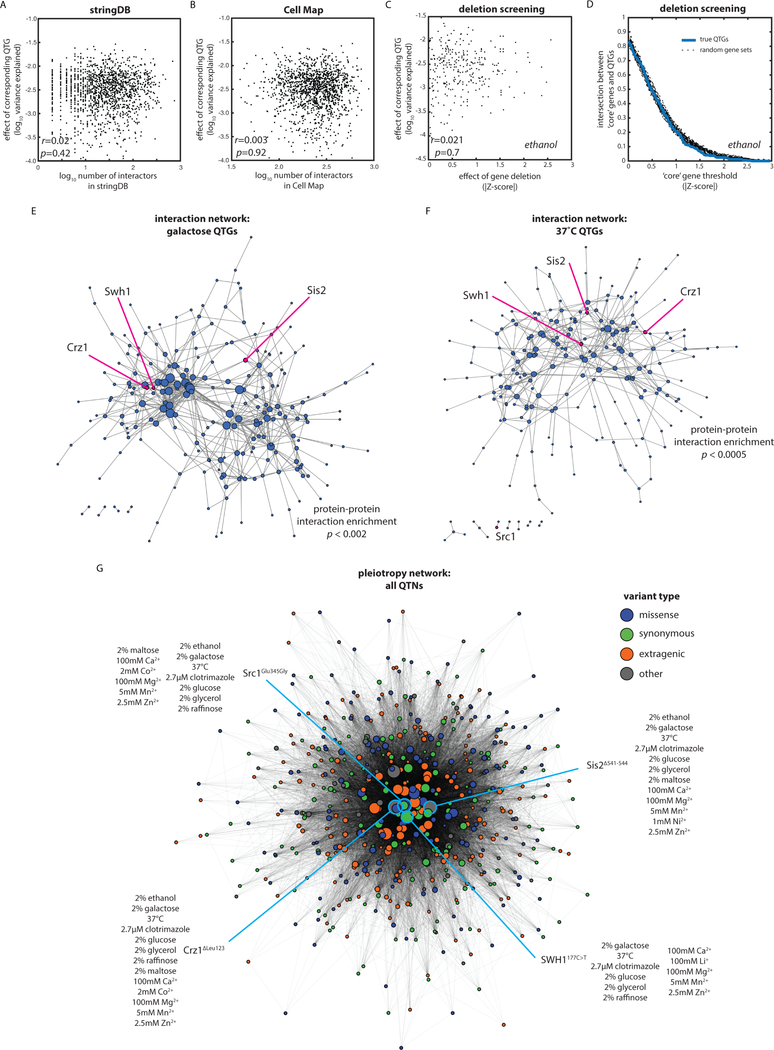
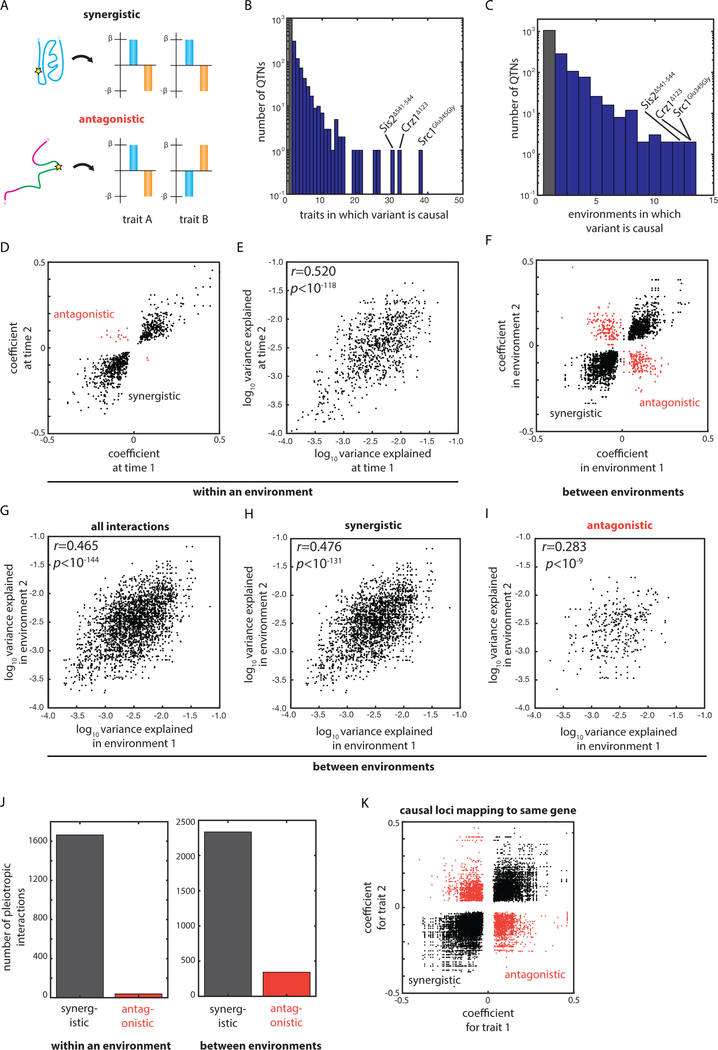
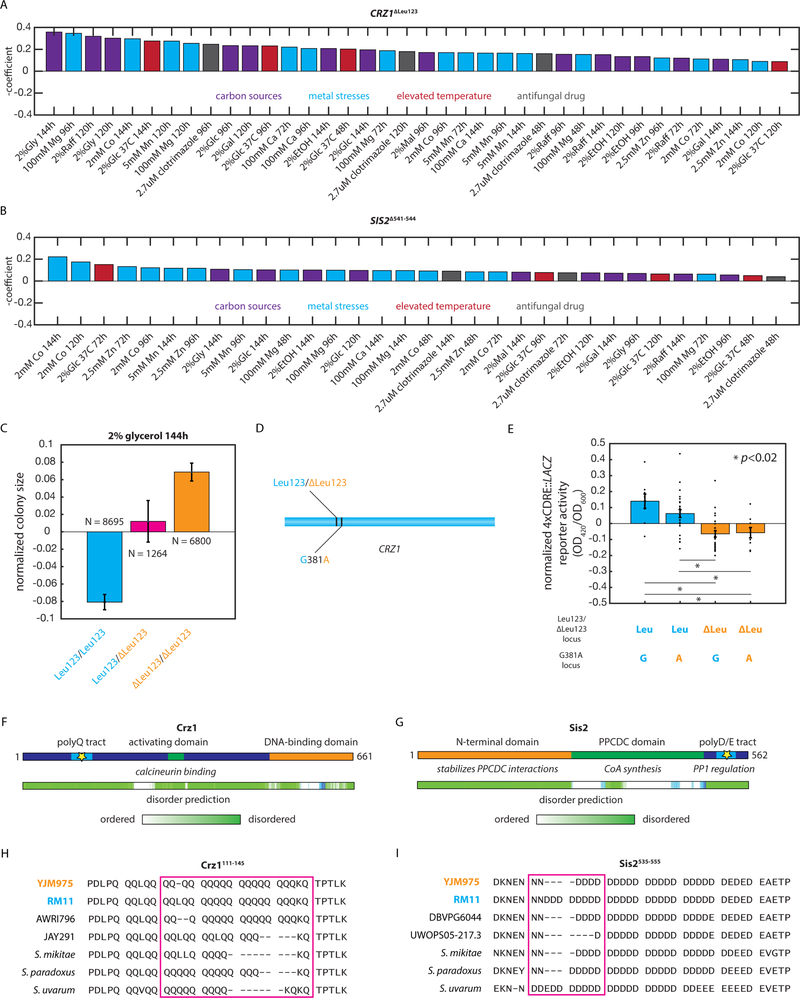
The statistical complexity of heredity has long been evident, but its molecular origins remain elusive. To investigate, we charted 90 comprehensive genotype-to-phenotype maps in a large population of wild diploid yeast. In contrast to long-standing assumptions, all types of genetic variation contributed similarly to phenotype. Causal synonymous and regulatory variants exhibited distinct molecular signatures, as did nonlinearities in heterozygote fitness that likely contribute to hybrid vigor. Highly pleiotropic variants altered disordered sequences within signaling hubs, and their effects correlated across environments-even when antagonistic-suggesting that large fitness gains bring concomitant costs. Natural genetic networks defined by the causal loci differed from those determined by precise gene deletions or protein-protein interactions. Finally, we found that traits that would appear omnigenic in less powered studies do in fact have finite genetic determinants. Integrating these molecular principles will be crucial as genome reading and writing become routine in research, industry, and medicine.
遗传的统计复杂性早已显而易见,但它的分子起源仍然难以捉摸。为了研究这一现象,我们在一个大型野生二倍体酵母群体中绘制了 90 张综合基因型到表型图谱。与长期以来的假设相反,所有类型的遗传变异都同样对表型有贡献。因果同义变体和调控变体表现出不同的分子特征,杂合子适应性的非线性也同样如此,这可能有助于杂种优势。高度多效性变体改变了信号枢纽内的无序序列,并且它们的效应在不同环境中具有相关性——即使是相互拮抗的——表明大的适应度增益伴随着相应的代价。由因果位点定义的自然遗传网络与由精确基因缺失或蛋白质-蛋白质相互作用决定的网络不同。最后,我们发现,在功能较弱的研究中看似全基因组遗传的性状实际上确实有有限的遗传决定因素。随着基因组阅读和编写在研究、工业和医学中变得常规化,整合这些分子原理将是至关重要的。