Giulini Marco, Potestio Raffaello
Physics Department, University of Trento, via Sommarive 14, 38123, Trento, Italy.
INFN-TIFPA, Trento Institute for Fundamental Physics and Applications, 38123 Trento, Italy.
Interface Focus. 2019 Jun 6;9(3):20190003. doi: 10.1098/rsfs.2019.0003. Epub 2019 Apr 19.
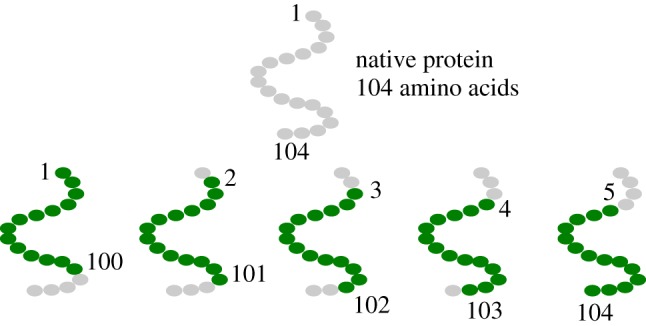
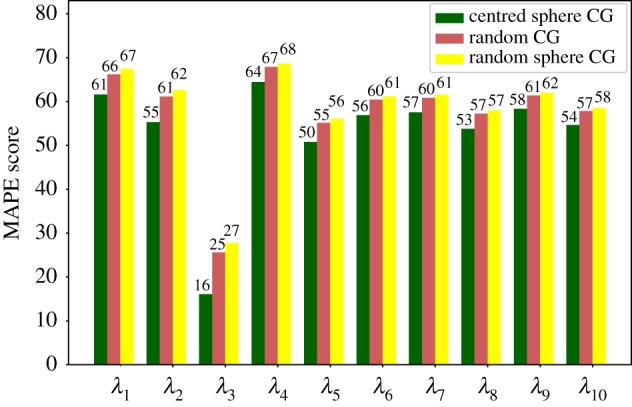
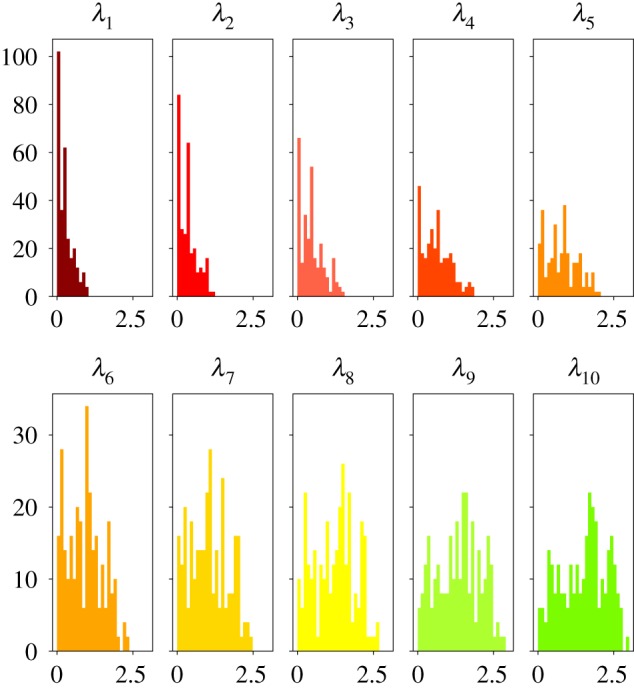
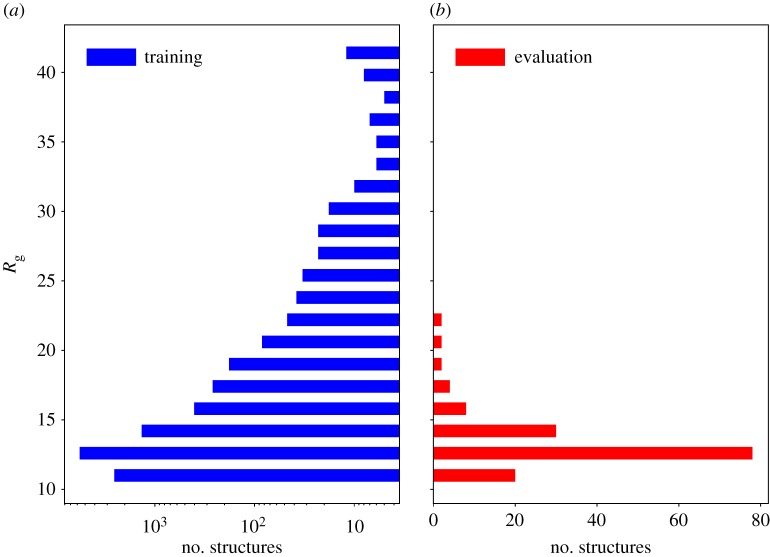
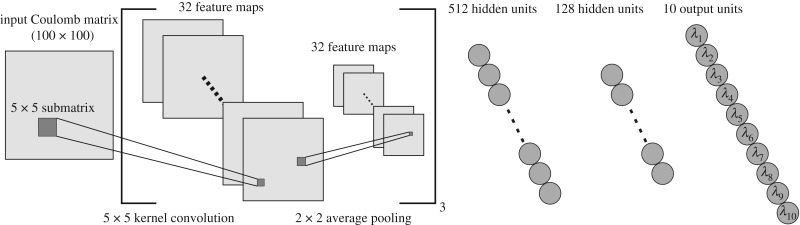
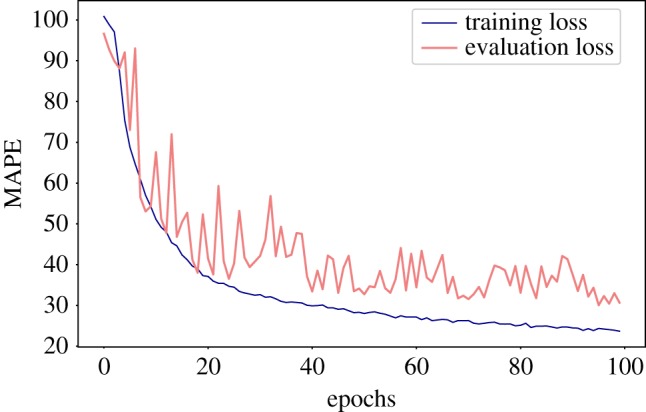
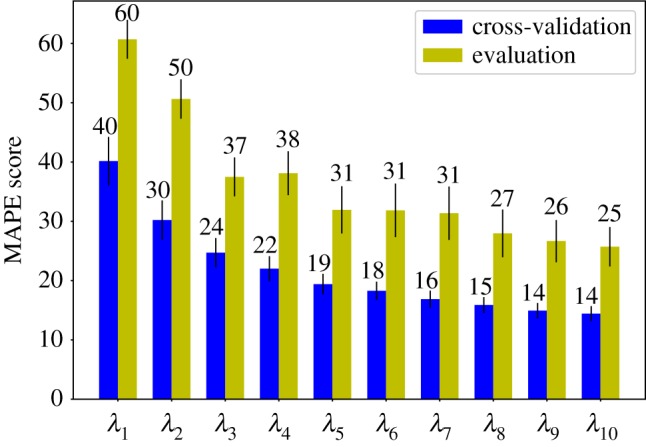
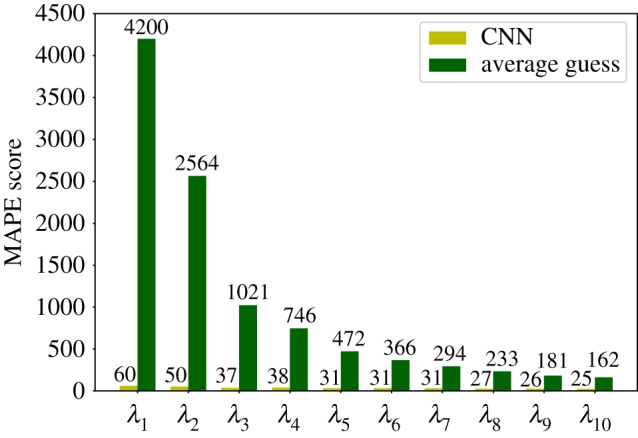
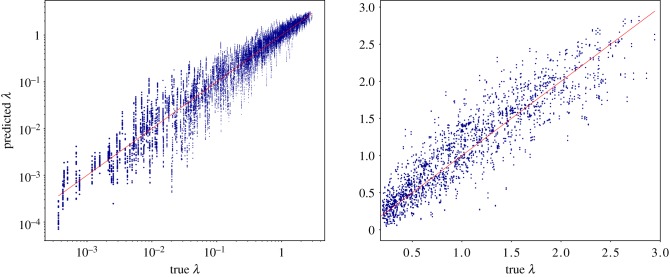
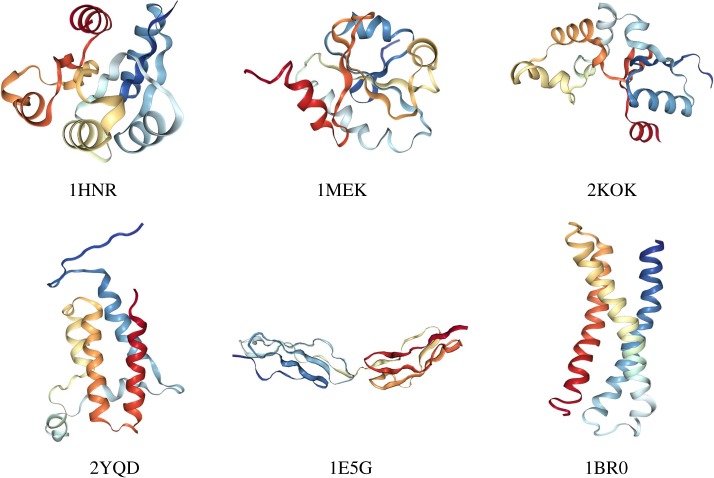
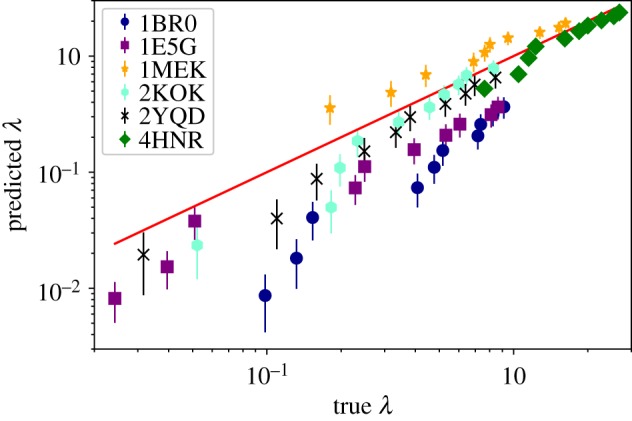
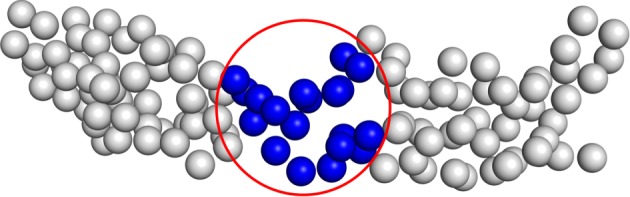
Deep learning (DL) algorithms hold great promise for applications in the field of computational biophysics. In fact, the vast amount of available molecular structures, as well as their notable complexity, constitutes an ideal context in which DL-based approaches can be profitably employed. To express the full potential of these techniques, though, it is a prerequisite to express the information contained in a molecule's atomic positions and distances in a set of input quantities that the network can process. Many of the molecular descriptors devised so far are effective and manageable for relatively small structures, but become complex and cumbersome for larger ones. Furthermore, most of them are defined locally, a feature that could represent a limit for those applications where global properties are of interest. Here, we build a DL architecture capable of predicting non-trivial and intrinsically global quantities, that is, the eigenvalues of a protein's lowest-energy fluctuation modes. This application represents a first, relatively simple test bed for the development of a neural network approach to the quantitative analysis of protein structures, and demonstrates unexpected use in the identification of mechanically relevant regions of the molecule.
深度学习(DL)算法在计算生物物理学领域的应用前景广阔。事实上,大量可用的分子结构及其显著的复杂性,构成了一个理想的环境,基于DL的方法可以在其中得到有效应用。然而,为了充分发挥这些技术的潜力,将分子原子位置和距离中包含的信息表示为网络可以处理的一组输入量是一个先决条件。到目前为止设计的许多分子描述符对于相对较小的结构是有效且易于管理的,但对于较大的结构则变得复杂且繁琐。此外,它们中的大多数是局部定义的,这一特征对于那些关注全局性质的应用来说可能是一个限制。在这里,我们构建了一种DL架构,能够预测非平凡且本质上全局的量,即蛋白质最低能量波动模式的特征值。该应用代表了开发用于蛋白质结构定量分析的神经网络方法的第一个相对简单的测试平台,并展示了其在识别分子机械相关区域方面的意外用途。