Parasar Bibudha, Zhou Hao, Xiao Xieyue, Shi Qiaojuan, Brito Ilana L, Chang Pamela V
Department of Chemistry and Chemical Biology, Department of Microbiology, Meinig School of Biomedical Engineering, Center for Infection and Pathobiology, Cornell Institute of Host-Microbe Interactions & Disease, and Department of Microbiology and Immunology, Cornell University, Ithaca, New York 14853, United States.
ACS Cent Sci. 2019 May 22;5(5):867-873. doi: 10.1021/acscentsci.9b00147. Epub 2019 Apr 18.
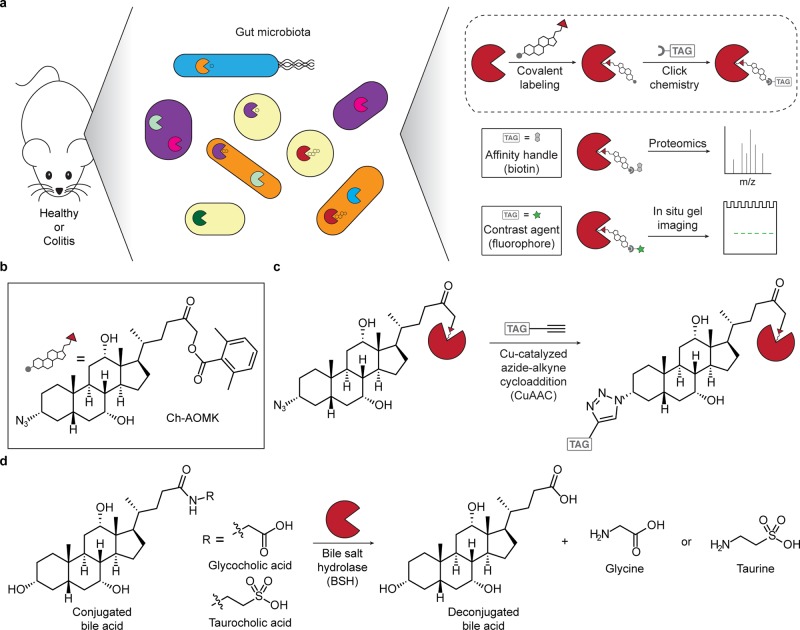
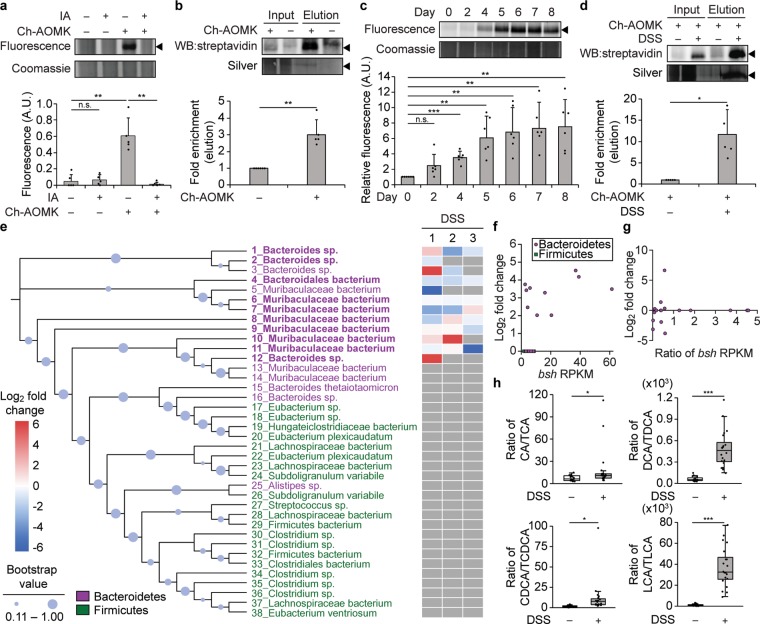
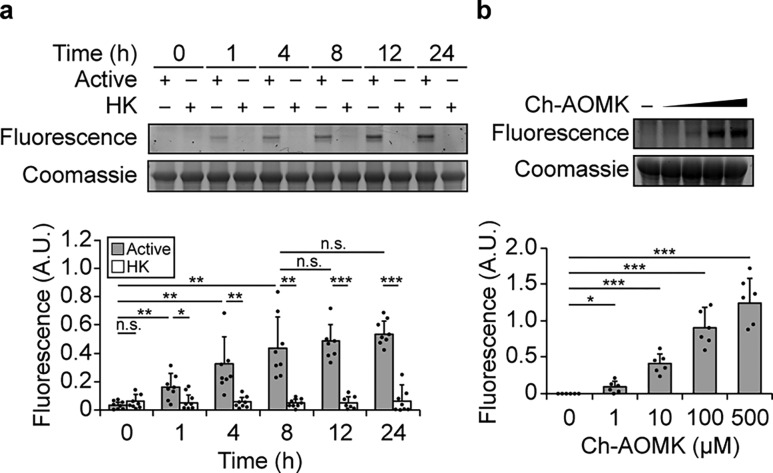
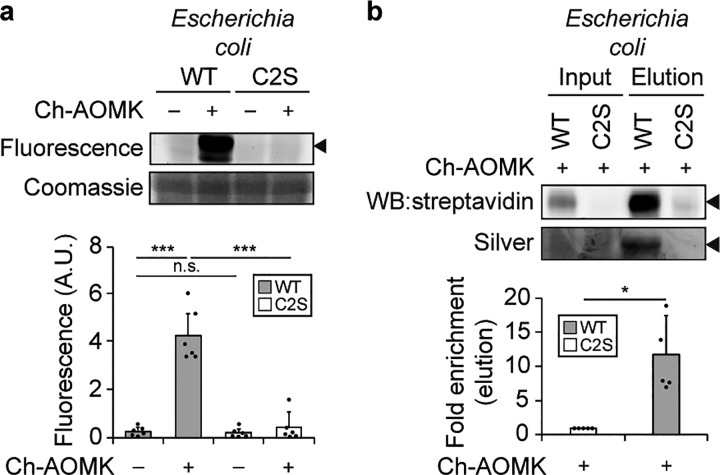
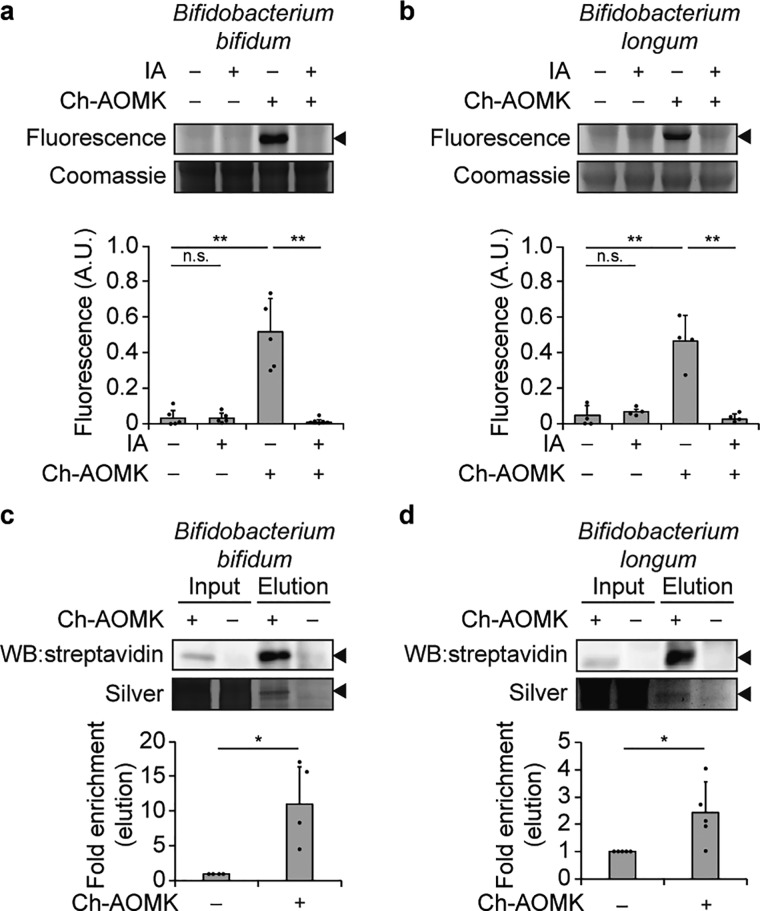
The metagenome of the gut microbiome encodes tremendous potential for biosynthesizing and transforming small-molecule metabolites through the activities of enzymes expressed by intestinal bacteria. Accordingly, elucidating this metabolic network is critical for understanding how the gut microbiota contributes to health and disease. Bile acids, which are first biosynthesized in the liver, are modified in the gut by enzymes expressed by commensal bacteria into secondary bile acids, which regulate myriad host processes, including lipid metabolism, glucose metabolism, and immune homeostasis. The gateway reaction of secondary bile acid biosynthesis is mediated by bile salt hydrolases (BSHs), bacterial cysteine hydrolases whose action precedes other bile acid modifications within the gut. To assess how changes in bile acid metabolism mediated by certain intestinal microbiota impact gut physiology and pathobiology, methods are needed to directly examine the activities of BSHs because they are master regulators of intestinal bile acid metabolism. Here, we developed chemoproteomic tools to profile changes in gut microbiome-associated BSH activity. We showed that these probes can label active BSHs in model microorganisms, including relevant gut anaerobes, and in mouse gut microbiomes. Using these tools, we identified altered BSH activities in a murine model of inflammatory bowel disease, in this case, colitis induced by dextran sodium sulfate, leading to changes in bile acid metabolism that could impact host metabolism and immunity. Importantly, our findings reveal that alterations in BSH enzymatic activities within the gut microbiome do not correlate with changes in gene abundance as determined by metagenomic sequencing, highlighting the utility of chemoproteomic approaches for interrogating the metabolic activities of the gut microbiota.
肠道微生物群的宏基因组通过肠道细菌表达的酶的活性,在生物合成和转化小分子代谢物方面具有巨大潜力。因此,阐明这一代谢网络对于理解肠道微生物群如何影响健康和疾病至关重要。胆汁酸首先在肝脏中生物合成,然后在肠道中被共生细菌表达的酶修饰为次级胆汁酸,次级胆汁酸调节多种宿主过程,包括脂质代谢、葡萄糖代谢和免疫稳态。次级胆汁酸生物合成的起始反应由胆汁盐水解酶(BSHs)介导,胆汁盐水解酶是一种细菌半胱氨酸水解酶,其作用先于肠道内其他胆汁酸修饰。为了评估某些肠道微生物群介导的胆汁酸代谢变化如何影响肠道生理和病理生物学,需要直接检测胆汁盐水解酶活性的方法,因为它们是肠道胆汁酸代谢的主要调节因子。在这里,我们开发了化学蛋白质组学工具来分析肠道微生物群相关胆汁盐水解酶活性的变化。我们表明,这些探针可以标记模型微生物(包括相关肠道厌氧菌)和小鼠肠道微生物群中的活性胆汁盐水解酶。使用这些工具,我们在炎症性肠病小鼠模型(在这种情况下,由葡聚糖硫酸钠诱导的结肠炎)中鉴定出胆汁盐水解酶活性的改变,这导致胆汁酸代谢的变化,进而可能影响宿主代谢和免疫。重要的是,我们的研究结果表明,肠道微生物群中胆汁盐水解酶活性的改变与宏基因组测序确定的基因丰度变化无关,这突出了化学蛋白质组学方法在研究肠道微生物群代谢活性方面的实用性。