Hagey Jill V, Bhatnagar Srijak, Heguy Jennifer M, Karle Betsy M, Price Patricia L, Meyer Deanne, Maga Elizabeth A
Department of Animal Science, University of California, Davis, Davis, CA, United States.
Geomicrobiology Group, Department of Biological Sciences, University of Calgary, Calgary, AB, Canada.
Front Microbiol. 2019 May 16;10:1093. doi: 10.3389/fmicb.2019.01093. eCollection 2019.
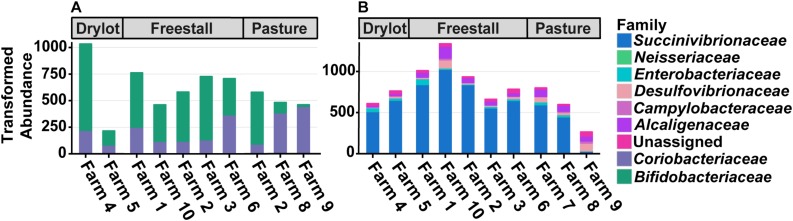
Improved sequencing and analytical techniques allow for better resolution of microbial communities; however, the agriculture field lacks an updated analysis surveying the fecal microbial populations of dairy cattle in California. This study is a large-scale survey to determine the composition of the bacterial community present in the feces of lactating dairy cattle on commercial dairy operations. For the study, 10 dairy farms across northern and central California representing a variety of feeding and management systems were enrolled. The farms represented three typical housing types including five freestall, two drylot and three pasture-based management systems. Fresh feces were collected from 15 randomly selected cows on each farm and analyzed using 16S rRNA gene amplicon sequencing. This study found that housing type, individual farm, and dietary components significantly affected the alpha diversity of the fecal microbiota. While only one Operational Taxonomic Unit (OTU) was common among all the sampled individuals, 15 bacterial families and 27 genera were shared among 95% of samples. The ratio of the families to was significantly different between housing types and farms with pasture fed animals having a higher relative abundance of . A majority of samples were positive for at least one OTU assigned to and 31% of samples contained OTUs assigned to . However, the relative abundance of both taxa was <0.1%. The microbial composition displays individual farm specific signatures, but housing type plays a role. These data provide insights into the composition of the core fecal microbiota of commercial dairy cows in California and will further generate hypotheses for strategies to manipulate the microbiome of cattle.
改进的测序和分析技术有助于更好地解析微生物群落;然而,农业领域缺乏一项针对加利福尼亚州奶牛粪便微生物种群的最新分析调查。本研究是一项大规模调查,旨在确定商业奶牛场泌乳奶牛粪便中细菌群落的组成。在该研究中,招募了加利福尼亚州北部和中部的10个奶牛场,这些奶牛场代表了各种饲养和管理系统。这些农场代表了三种典型的饲养类型,包括五个散栏式、两个干栏式和三个基于牧场的管理系统。从每个农场随机挑选的15头奶牛收集新鲜粪便,并使用16S rRNA基因扩增子测序进行分析。本研究发现,饲养类型、单个农场和饮食成分显著影响粪便微生物群的α多样性。虽然在所有采样个体中只有一个操作分类单元(OTU)是常见的,但在95%的样本中共有15个细菌科和27个属。不同饲养类型和农场之间,科与科的比例存在显著差异,以牧场饲养的动物中相对丰度较高。大多数样本至少有一个分配给的OTU呈阳性,31%的样本包含分配给的OTU。然而,这两个分类群的相对丰度均<0.1%。微生物组成显示出各个农场的特定特征,但饲养类型也起到了一定作用。这些数据为加利福尼亚州商业奶牛核心粪便微生物群的组成提供了见解,并将进一步为操纵奶牛微生物组的策略生成假设。