Gomig Talita Helen Bombardelli, Cavalli Iglenir João, Souza Ricardo Lehtonen Rodrigues de, Lucena Aline Castro Rodrigues, Batista Michel, Machado Kelly Cavalcanti, Marchini Fabricio Klerynton, Marchi Fabio Albuquerque, Lima Rubens Silveira, Urban Cícero de Andrade, Cavalli Luciane Regina, Ribeiro Enilze Maria de Souza Fonseca
Genetics Department, Federal University of Parana, Curitiba, Brazil.
Functional Genomics Laboratory, Carlos Chagas Institute, Fiocruz, Curitiba, Parana, Brazil.
Data Brief. 2019 Jun 10;25:104125. doi: 10.1016/j.dib.2019.104125. eCollection 2019 Aug.
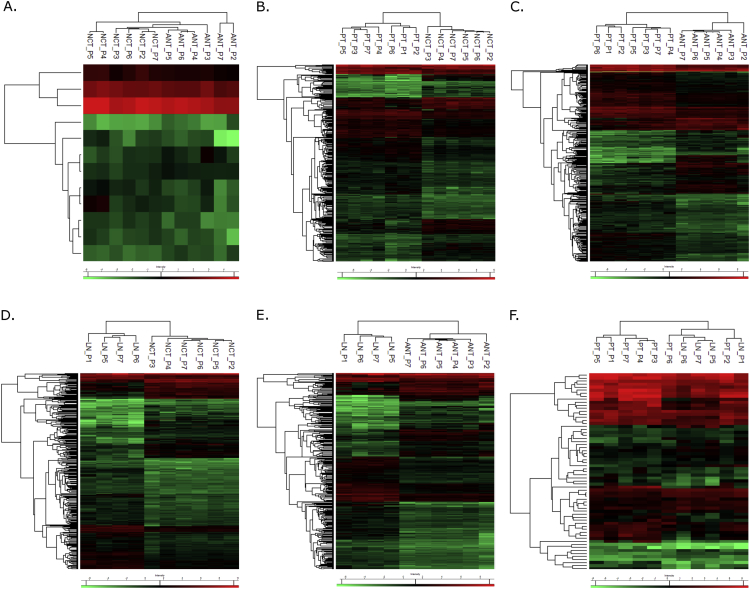
Data present here describe a comparative proteomic analysis among the malignant [primary breast tumor (PT) and axillary metastatic lymph nodes (LN)], and the non-tumor [contralateral (NCT) and adjacent (ANT)] breast tissues. Protein identification and quantification were performed through label-free mass spectrometry using a nano-liquid chromatography coupled to an electrospray ionization-mass spectrometry (nLC-ESI-MS/MS). The mass spectrometry proteomic data have been deposited to the ProteomeXchange Consortium via PRIDE partner repository with the dataset identifier PXD012431. A total of 462 differentially expressed proteins was identified among these tissues and was analyzed in six groups' comparisons (named NCTxANT, PTxNCT, PTxANT, LNxNCT, LNxANT and PTxLN). Proteins at 1.5 log2 fold change were submitted to the Ingenuity Pathway Analysis (IPA) software version 2.3 (QIAGEN Inc.) to identify biological pathways, disease and function annotation, and interaction networks related to cancer biology. The detailed data present here provides information about the proteome alterations and their role on breast tumorigenesis. This information can lead to novel biological insights on cancer research. For further interpretation of these data, please see our research article 'Quantitative label-free mass spectrometry using contralateral and adjacent breast tissues reveal differentially expressed proteins and their predicted impacts on pathways and cellular functions in breast cancer' [2].
本文所呈现的数据描述了恶性组织[原发性乳腺肿瘤(PT)和腋窝转移性淋巴结(LN)]与非肿瘤组织[对侧(NCT)和相邻(ANT)乳腺组织]之间的比较蛋白质组学分析。通过使用纳升液相色谱与电喷雾电离质谱联用(nLC-ESI-MS/MS)的无标记质谱法进行蛋白质鉴定和定量。质谱蛋白质组学数据已通过PRIDE合作伙伴库存入蛋白质组交换联盟,数据集标识符为PXD012431。在这些组织中总共鉴定出462种差异表达蛋白质,并在六组比较中进行了分析(命名为NCTxANT、PTxNCT、PTxANT、LNxNCT、LNxANT和PTxLN)。将1.5 log2倍变化的蛋白质提交给Ingenuity Pathway Analysis(IPA)软件版本2.3(QIAGEN公司),以识别与癌症生物学相关的生物途径、疾病和功能注释以及相互作用网络。本文所呈现的详细数据提供了有关蛋白质组改变及其在乳腺肿瘤发生中作用的信息。这些信息可带来癌症研究方面的新生物学见解。有关这些数据的进一步解读,请参阅我们的研究文章《使用对侧和相邻乳腺组织的无标记定量质谱法揭示差异表达蛋白质及其对乳腺癌途径和细胞功能的预测影响》[2]。