Yang Xiaodi, Yang Shiping, Li Qinmengge, Wuchty Stefan, Zhang Ziding
State Key Laboratory of Agrobiotechnology, College of Biological Sciences, China Agricultural University, Beijing 100193, China.
State Key Laboratory of Plant Physiology and Biochemistry, College of Biological Sciences, China Agricultural University, Beijing 100193, China.
Comput Struct Biotechnol J. 2019 Dec 26;18:153-161. doi: 10.1016/j.csbj.2019.12.005. eCollection 2020.
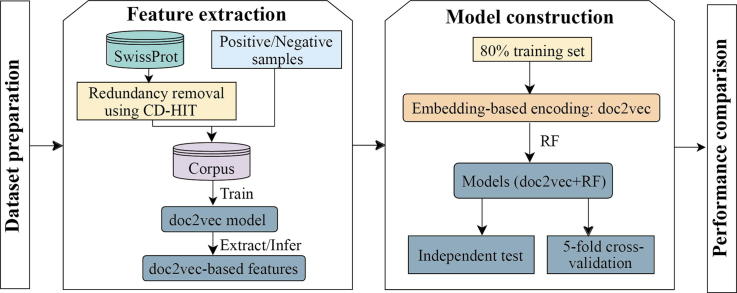
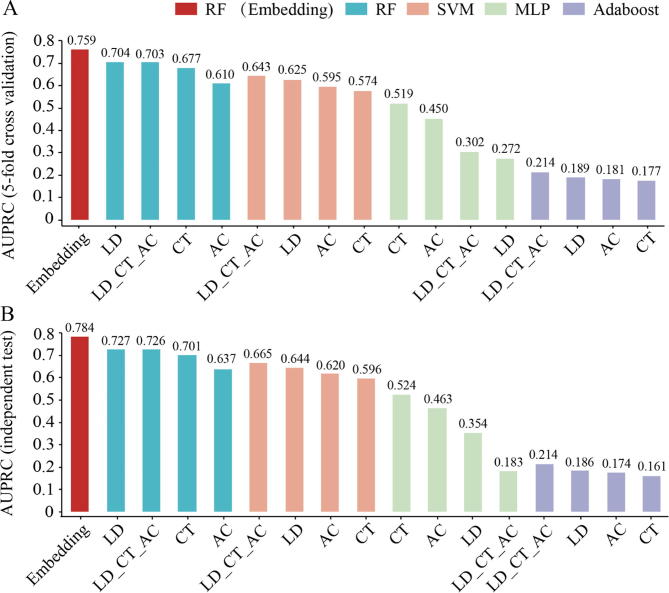
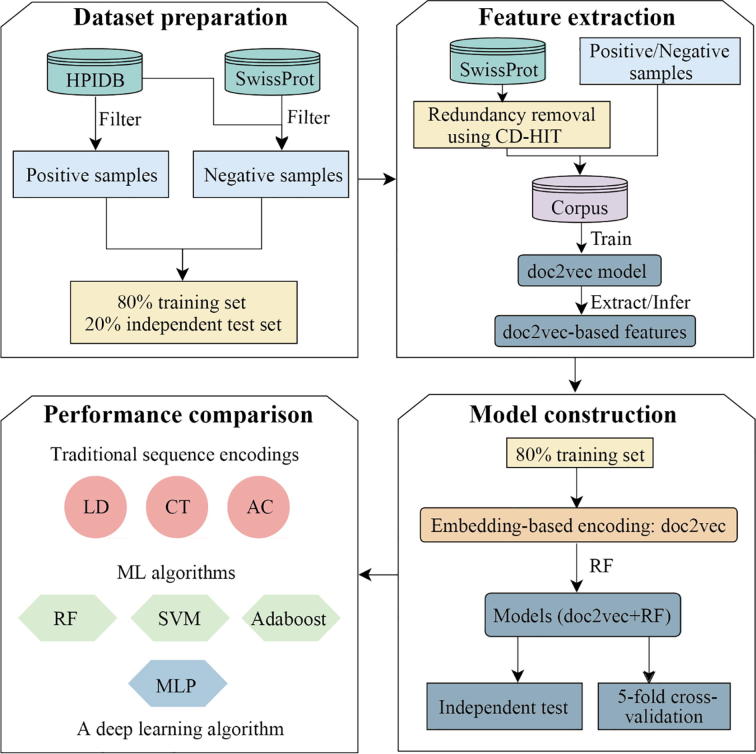
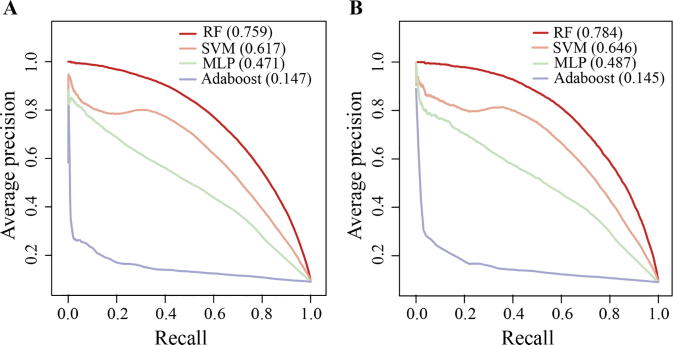
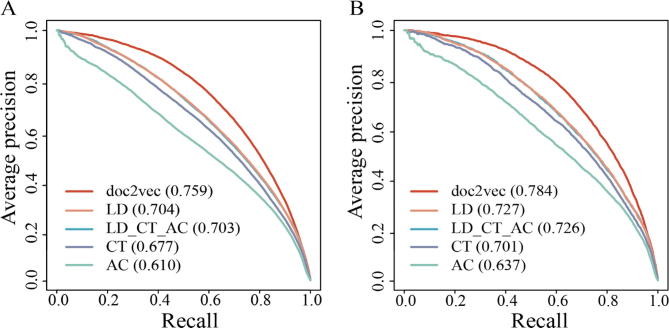
The identification of human-virus protein-protein interactions (PPIs) is an essential and challenging research topic, potentially providing a mechanistic understanding of viral infection. Given that the experimental determination of human-virus PPIs is time-consuming and labor-intensive, computational methods are playing an important role in providing testable hypotheses, complementing the determination of large-scale interactome between species. In this work, we applied an unsupervised sequence embedding technique (doc2vec) to represent protein sequences as rich feature vectors of low dimensionality. Training a Random Forest (RF) classifier through a training dataset that covers known PPIs between human and all viruses, we obtained excellent predictive accuracy outperforming various combinations of machine learning algorithms and commonly-used sequence encoding schemes. Rigorous comparison with three existing human-virus PPI prediction methods, our proposed computational framework further provided very competitive and promising performance, suggesting that the doc2vec encoding scheme effectively captures context information of protein sequences, pertaining to corresponding protein-protein interactions. Our approach is freely accessible through our web server as part of our host-pathogen PPI prediction platform (http://zzdlab.com/InterSPPI/). Taken together, we hope the current work not only contributes a useful predictor to accelerate the exploration of human-virus PPIs, but also provides some meaningful insights into human-virus relationships.
鉴定人类与病毒的蛋白质-蛋白质相互作用(PPI)是一个至关重要且具有挑战性的研究课题,它有可能为病毒感染提供机制性的理解。鉴于通过实验确定人类与病毒的PPI既耗时又费力,计算方法在提供可测试的假设方面发挥着重要作用,对物种间大规模相互作用组的确定起到补充作用。在这项工作中,我们应用了一种无监督序列嵌入技术(doc2vec),将蛋白质序列表示为低维的丰富特征向量。通过一个涵盖人类与所有病毒之间已知PPI的训练数据集训练随机森林(RF)分类器,我们获得了出色的预测准确率,优于各种机器学习算法和常用序列编码方案的组合。与三种现有的人类与病毒PPI预测方法进行严格比较,我们提出的计算框架进一步展现出极具竞争力和前景的性能,这表明doc2vec编码方案有效地捕捉了与相应蛋白质-蛋白质相互作用相关的蛋白质序列上下文信息。我们的方法可通过我们的网络服务器免费获取,作为我们宿主-病原体PPI预测平台(http://zzdlab.com/InterSPPI/)的一部分。综上所述,我们希望当前的工作不仅能为加速人类与病毒PPI的探索贡献一个有用的预测工具,还能为人类与病毒的关系提供一些有意义的见解。