Lee Jae-Rin, Lee Jong-Yoon, Kim Hyun-Ji, Hahn Myong-Joon, Kang Jong-Sun, Cho Hana
Department of Molecular Cell Biology, Sungkyunkwan University, Suwon, Korea.
Single Cell Network Research Center, Sungkyunkwan University, Suwon, Korea.
Exp Mol Med. 2019 Jul 17;51(7):1-11. doi: 10.1038/s12276-019-0279-2.
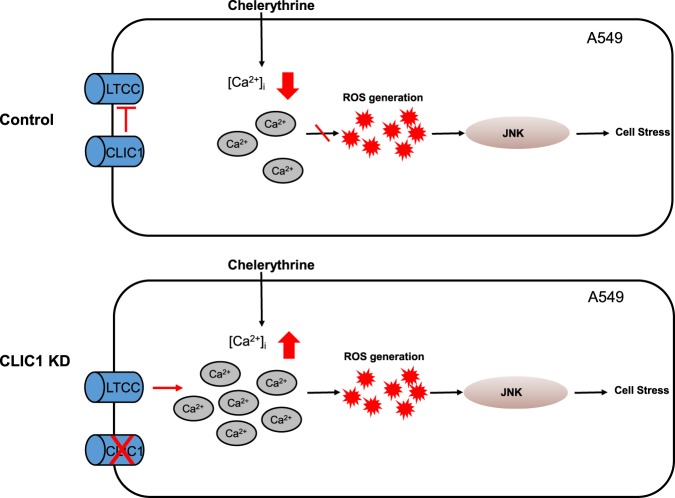
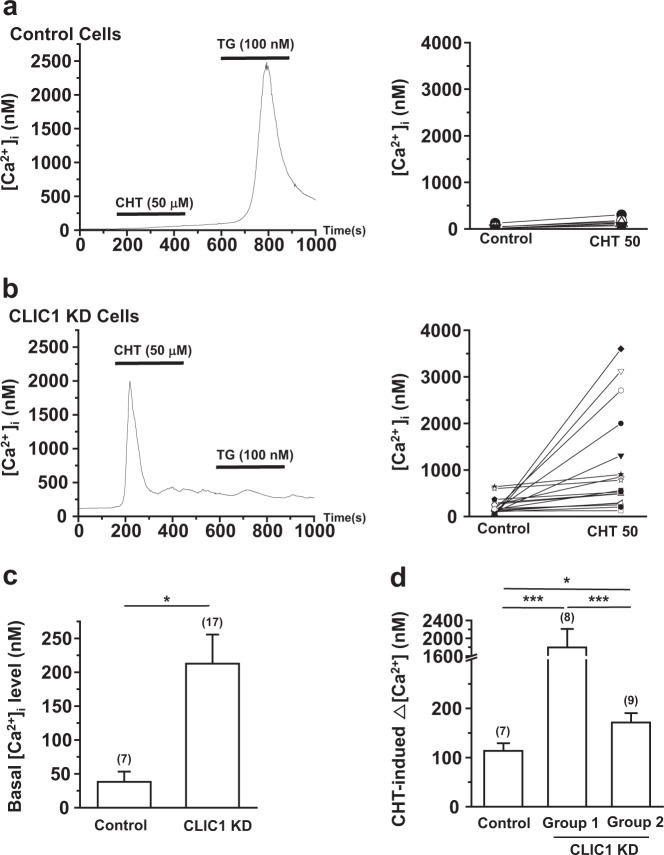
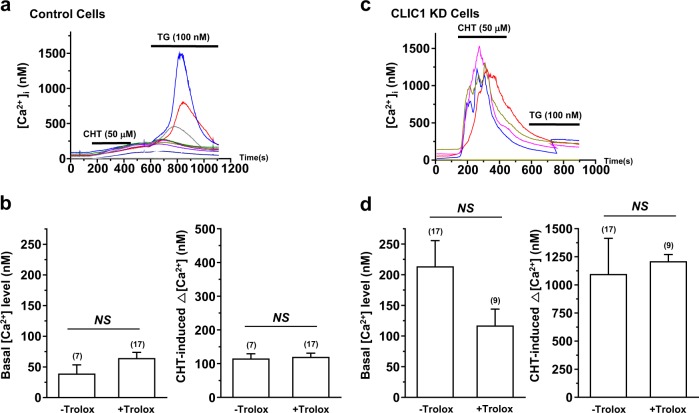
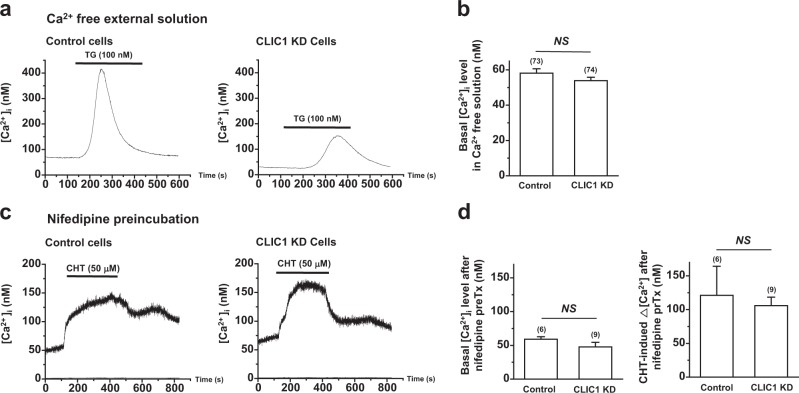
Chloride intracellular channel 1 (CLIC1) is a promising therapeutic target in cancer due to its intrinsic characteristics; it is overexpressed in specific tumor types and its localization changes from cytosolic to surface membrane depending on activities and cell cycle progression. Ca and reactive oxygen species (ROS) are critical signaling molecules that modulate diverse cellular functions, including cell death. In this study, we investigated the function of CLIC1 in Ca and ROS signaling in A549 human lung cancer cells. Depletion of CLIC1 via shRNAs in A549 cells increased DNA double-strand breaks both under control conditions and under treatment with the putative anticancer agent chelerythrine, accompanied by a concomitant increase in the p-JNK level. CLIC1 knockdown greatly increased basal ROS levels, an effect prevented by BAPTA-AM, an intracellular calcium chelator. Intracellular Ca measurements clearly showed that CLIC1 knockdown significantly increased chelerythrine-induced Ca signaling as well as the basal Ca level in A549 cells compared to these levels in control cells. Suppression of extracellular Ca restored the basal Ca level in CLIC1-knockdown A549 cells relative to that in control cells, implying that CLIC1 regulates [Ca] through Ca entry across the plasma membrane. Consistent with this finding, the L-type Ca channel (LTCC) blocker nifedipine reduced the basal Ca level in CLIC1 knockdown cells to that in control cells. Taken together, our results demonstrate that CLIC1 knockdown induces an increase in the intracellular Ca level via LTCC, which then triggers excessive ROS production and consequent JNK activation. Thus, CLIC1 is a key regulator of Ca signaling in the control of cancer cell survival.
氯离子细胞内通道1(CLIC1)因其内在特性而成为癌症治疗中一个有前景的靶点;它在特定肿瘤类型中过度表达,并且其定位会根据细胞活性和细胞周期进程从胞质溶胶转变为细胞膜表面。钙和活性氧(ROS)是调节多种细胞功能(包括细胞死亡)的关键信号分子。在本研究中,我们调查了CLIC1在A549人肺癌细胞中钙和ROS信号传导中的功能。通过短发夹RNA(shRNA)使A549细胞中的CLIC1缺失,在对照条件下以及在用推定的抗癌剂白屈菜红碱处理时,均增加了DNA双链断裂,同时伴随着p-JNK水平的相应增加。CLIC1基因敲低显著增加了基础ROS水平,而细胞内钙螯合剂BAPTA-AM可阻止这种效应。细胞内钙测量清楚地表明,与对照细胞相比,CLIC1基因敲低显著增加了白屈菜红碱诱导的A549细胞中的钙信号以及基础钙水平。抑制细胞外钙可使CLIC1基因敲低的A549细胞中的基础钙水平相对于对照细胞恢复到正常水平,这意味着CLIC1通过跨质膜的钙内流来调节[Ca]。与此发现一致,L型钙通道(LTCC)阻滞剂硝苯地平将CLIC1基因敲低细胞中的基础钙水平降低至对照细胞中的水平。综上所述,我们的结果表明,CLIC1基因敲低通过LTCC诱导细胞内钙水平升高,进而触发过量的ROS产生并随之激活JNK。因此,CLIC1是控制癌细胞存活中钙信号传导的关键调节因子。