Department of Biostatistics and Bioinformatics, H. Lee Moffitt Cancer Center and Research Institute, Tampa, FL, 33612, USA.
Department of Immunology, H. Lee Moffitt Cancer Center and Research Institute, Tampa, FL, 33612, USA.
BMC Cancer. 2019 Jul 19;19(1):715. doi: 10.1186/s12885-019-5927-3.
The rapid development of single-cell RNA sequencing (scRNA-seq) provides unprecedented opportunities to study the tumor ecosystem that involves a heterogeneous mixture of cell types. However, the majority of previous and current studies related to translational and molecular oncology have only focused on the bulk tumor and there is a wealth of gene expression data accumulated with matched clinical outcomes.
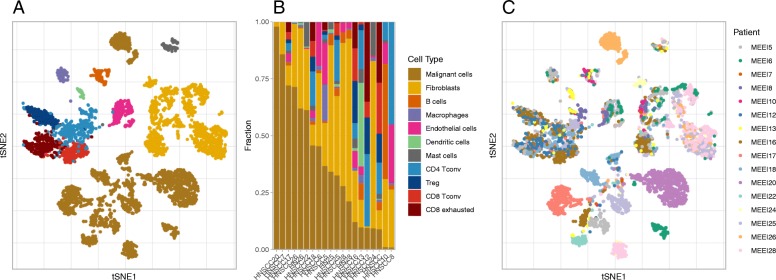
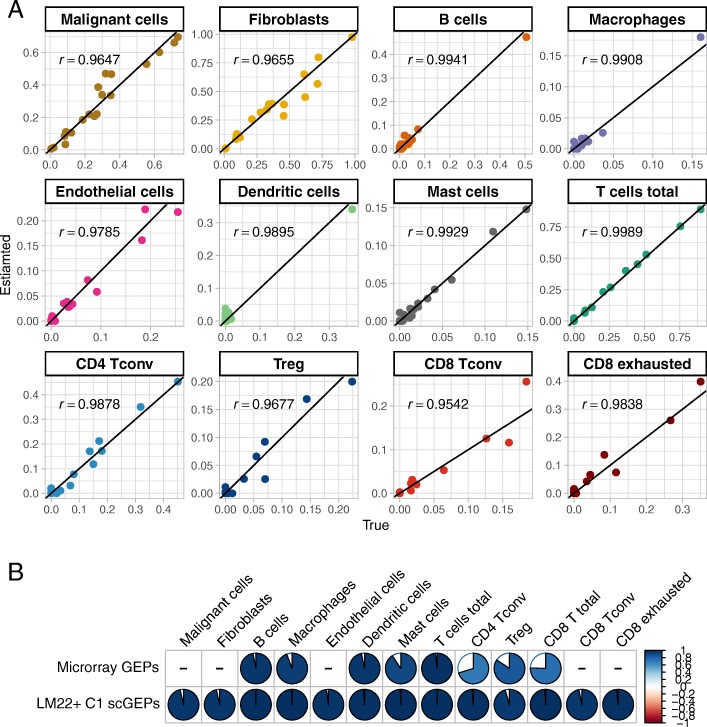
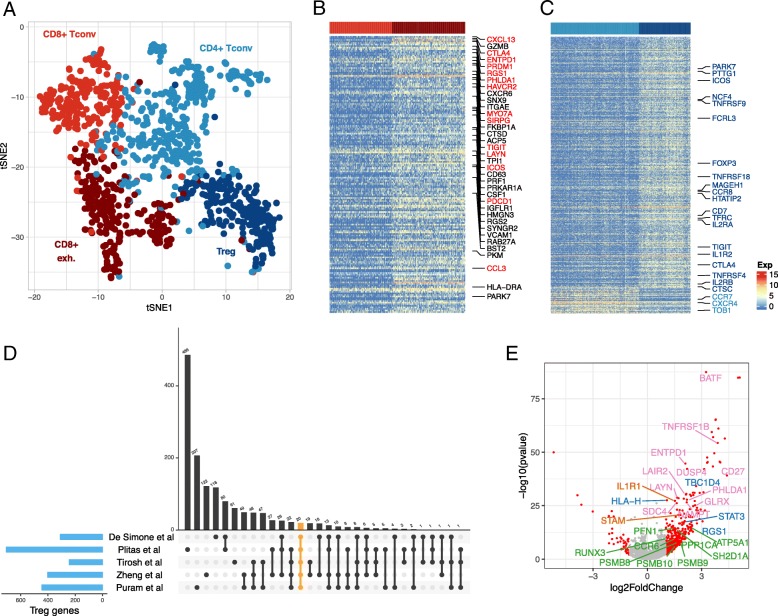
In this paper, we introduce a scheme for characterizing cell compositions from bulk tumor gene expression by integrating signatures learned from scRNA-seq data. We derived the reference expression matrix to each cell type based on cell subpopulations identified in head and neck cancer dataset. Our results suggest that scRNA-Seq-derived reference matrix outperforms the existing gene panel and reference matrix with respect to distinguishing immune cell subtypes.
Findings and resources created from this study enable future and secondary analysis of tumor RNA mixtures in head and neck cancer for a more accurate cellular deconvolution, and can facilitate the profiling of the immune infiltration in other solid tumors due to the expression homogeneity observed in immune cells.
单细胞 RNA 测序(scRNA-seq)的快速发展为研究肿瘤生态系统提供了前所未有的机会,该系统涉及多种细胞类型的混合。然而,大多数先前和当前与转化和分子肿瘤学相关的研究仅集中在肿瘤的整体上,并且有大量与匹配的临床结果相关的基因表达数据积累。
在本文中,我们通过整合从 scRNA-seq 数据中学习到的特征,介绍了一种从肿瘤整体基因表达中描述细胞组成的方案。我们基于在头颈癌数据集中确定的细胞亚群,为每种细胞类型推导了参考表达矩阵。我们的结果表明,与现有基因面板和参考矩阵相比,scRNA-Seq 衍生的参考矩阵在区分免疫细胞亚型方面表现更好。
本研究的发现和创建的资源可以促进对头颈癌肿瘤 RNA 混合物的进一步分析,以实现更准确的细胞去卷积,并由于观察到免疫细胞中的表达均匀性,可促进其他实体肿瘤中免疫浸润的分析。