Basic Forestry and Proteomics Research Center, College of Life Science, Fujian Provincial Key Laboratory of Haixia Applied Plant Systems Biology, Fujian Agriculture and Forestry University, Fuzhou 350002, China.
Jilin Province Engineering Laboratory of Plant Genetic Improvement, College of Plant Science, Jilin University, 5333 Xi'an Road, Changchun 130062, China.
Cells. 2019 Jul 19;8(7):744. doi: 10.3390/cells8070744.
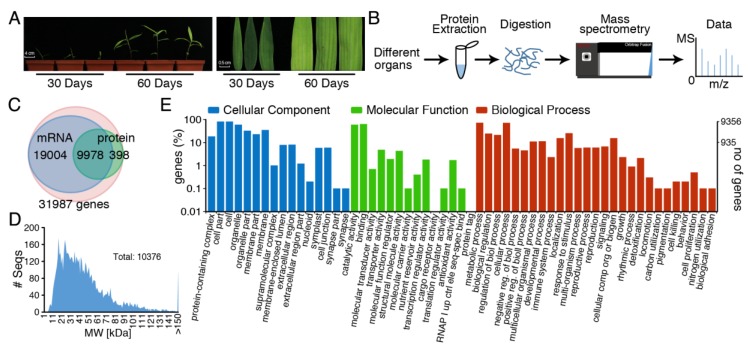
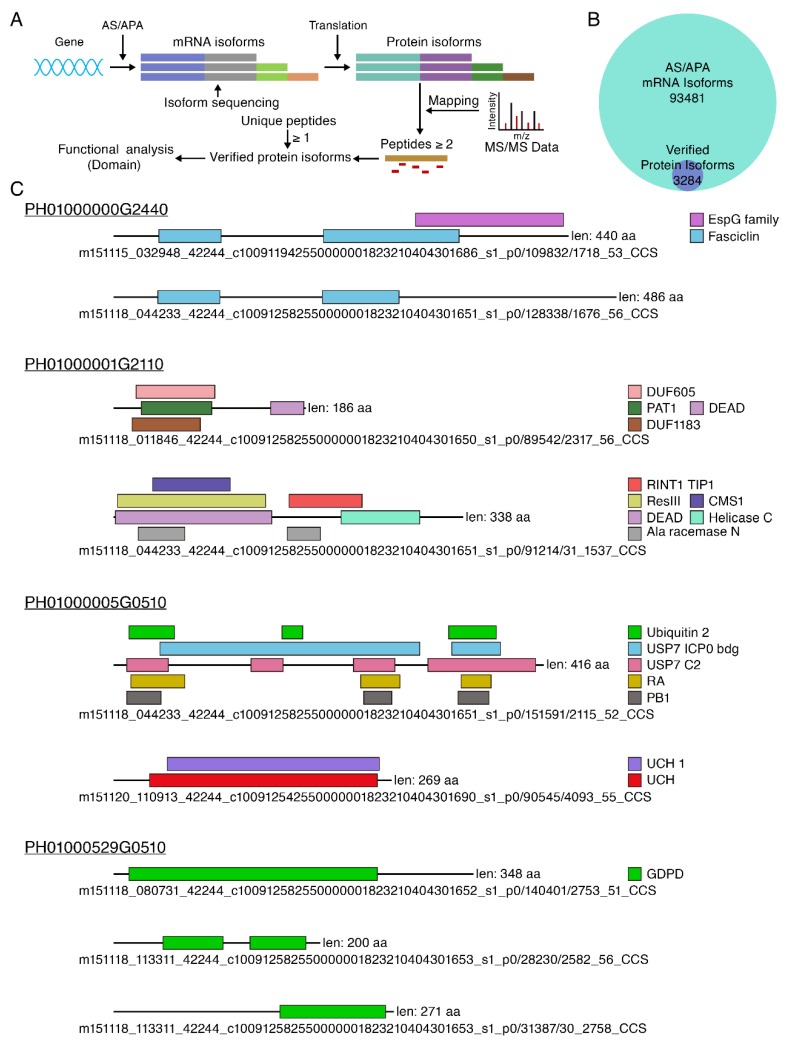
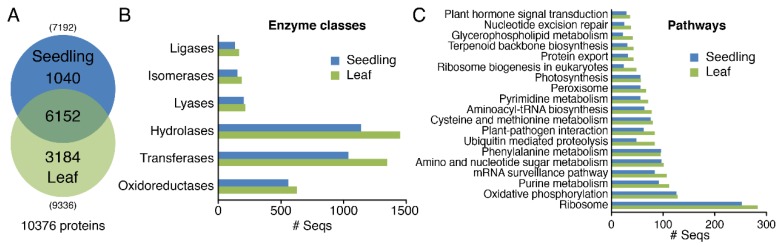
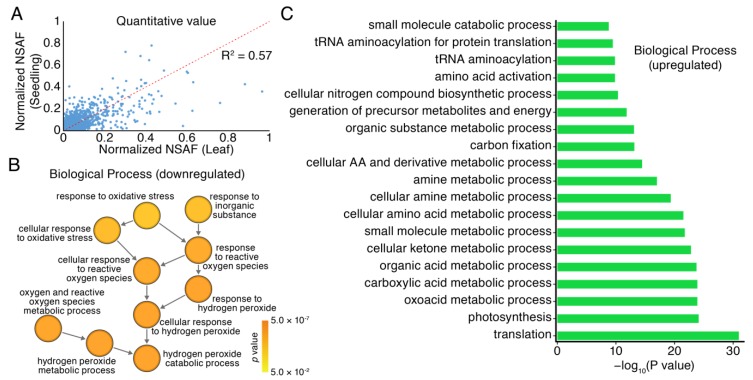
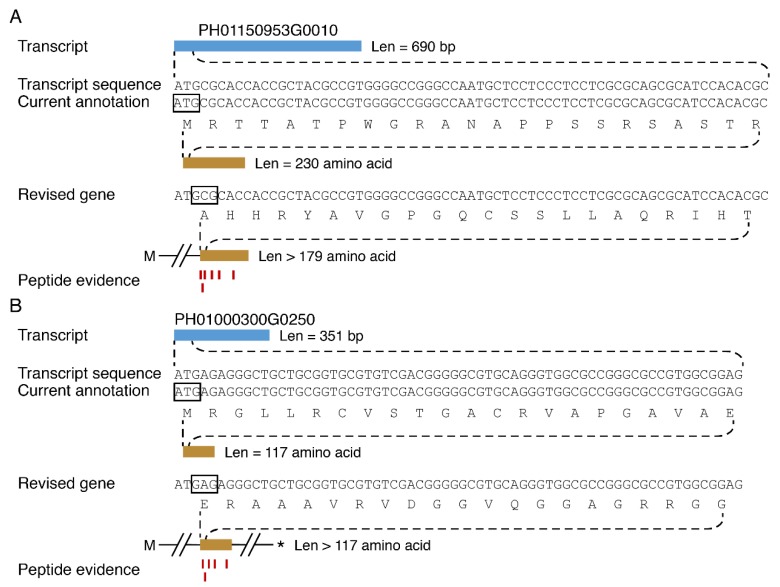
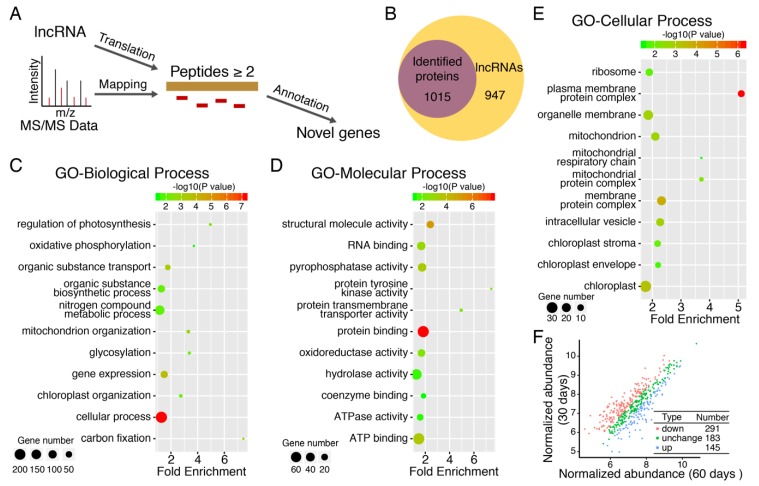
Moso bamboo is an important forest species with a variety of ecological, economic, and cultural values. However, the gene annotation information of moso bamboo is only based on the transcriptome sequencing, lacking the evidence of proteome. The lignification and fiber in moso bamboo leads to a difficulty in the extraction of protein using conventional methods, which seriously hinders research on the proteomics of moso bamboo. The purpose of this study is to establish efficient methods for extracting the total proteins from moso bamboo for following mass spectrometry-based quantitative proteome identification. Here, we have successfully established a set of efficient methods for extracting total proteins of moso bamboo followed by mass spectrometry-based label-free quantitative proteome identification, which further improved the protein annotation of moso bamboo genes. In this study, 10,376 predicted coding genes were confirmed by quantitative proteomics, accounting for 35.8% of all annotated protein-coding genes. Proteome analysis also revealed the protein-coding potential of 1015 predicted long noncoding RNA (lncRNA), accounting for 51.03% of annotated lncRNAs. Thus, mass spectrometry-based proteomics provides a reliable method for gene annotation. Especially, quantitative proteomics revealed the translation patterns of proteins in moso bamboo. In addition, the 3284 transcript isoforms from 2663 genes identified by Pacific BioSciences (PacBio) single-molecule real-time long-read isoform sequencing (Iso-Seq) was confirmed on the protein level by mass spectrometry. Furthermore, domain analysis of mass spectrometry-identified proteins encoded in the same genomic locus revealed variations in domain composition pointing towards a functional diversification of protein isoform. Finally, we found that part transcripts targeted by nonsense-mediated mRNA decay (NMD) could also be translated into proteins. In summary, proteomic analysis in this study improves the proteomics-assisted genome annotation of moso bamboo and is valuable to the large-scale research of functional genomics in moso bamboo. In summary, this study provided a theoretical basis and technical support for directional gene function analysis at the proteomics level in moso bamboo.
丛生竹是一种具有多种生态、经济和文化价值的重要森林物种。然而,丛生竹的基因注释信息仅基于转录组测序,缺乏蛋白质组的证据。丛生竹的木质化和纤维导致常规方法提取蛋白质变得困难,这严重阻碍了丛生竹蛋白质组学的研究。本研究旨在建立从丛生竹中提取总蛋白质的有效方法,用于后续基于质谱的定量蛋白质组鉴定。在这里,我们成功地建立了一套从丛生竹中提取总蛋白质的有效方法,用于基于质谱的无标记定量蛋白质组鉴定,进一步提高了丛生竹基因的注释。在这项研究中,通过定量蛋白质组学鉴定了 10376 个预测的编码基因,占所有注释的蛋白质编码基因的 35.8%。蛋白质组分析还揭示了 1015 个预测的长非编码 RNA(lncRNA)的蛋白质编码潜力,占注释的 lncRNA 的 51.03%。因此,基于质谱的蛋白质组学为基因注释提供了一种可靠的方法。特别是,定量蛋白质组学揭示了丛生竹中蛋白质的翻译模式。此外,通过 Pacific BioSciences(PacBio)单分子实时长读长读 isoform 测序(Iso-Seq)鉴定的 2663 个基因的 3284 个转录异构体在蛋白质水平上通过质谱得到了确认。此外,对同一基因组座上编码的质谱鉴定的蛋白质所包含的结构域分析表明,结构域组成的变化指向蛋白质同工型功能多样化。最后,我们发现部分受无意义介导的 mRNA 降解(NMD)靶向的转录本也可以被翻译为蛋白质。总之,本研究中的蛋白质组分析提高了丛生竹的蛋白质组辅助基因组注释,并为丛生竹大规模功能基因组学研究提供了有价值的信息。总之,本研究为丛生竹在蛋白质组学水平上进行定向基因功能分析提供了理论依据和技术支持。