a Department of Microbiology and Immunology, University of Texas Medical Branch , Galveston , USA.
b Department of Biochemistry and Molecular Biology, University of Texas Medical Branch , Galveston , USA.
Emerg Microbes Infect. 2019;8(1):1126-1138. doi: 10.1080/22221751.2019.1645572.
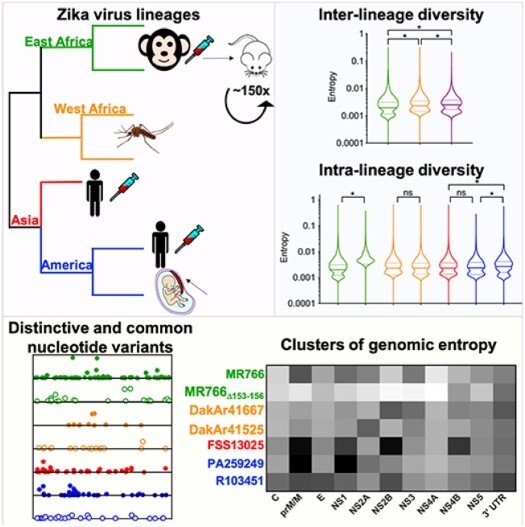
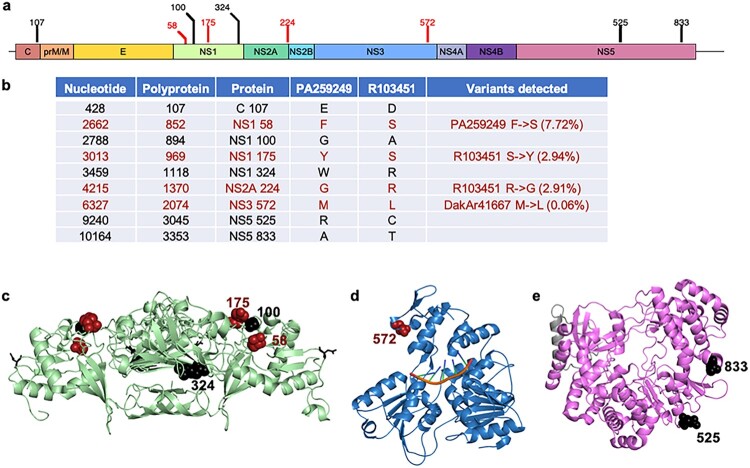
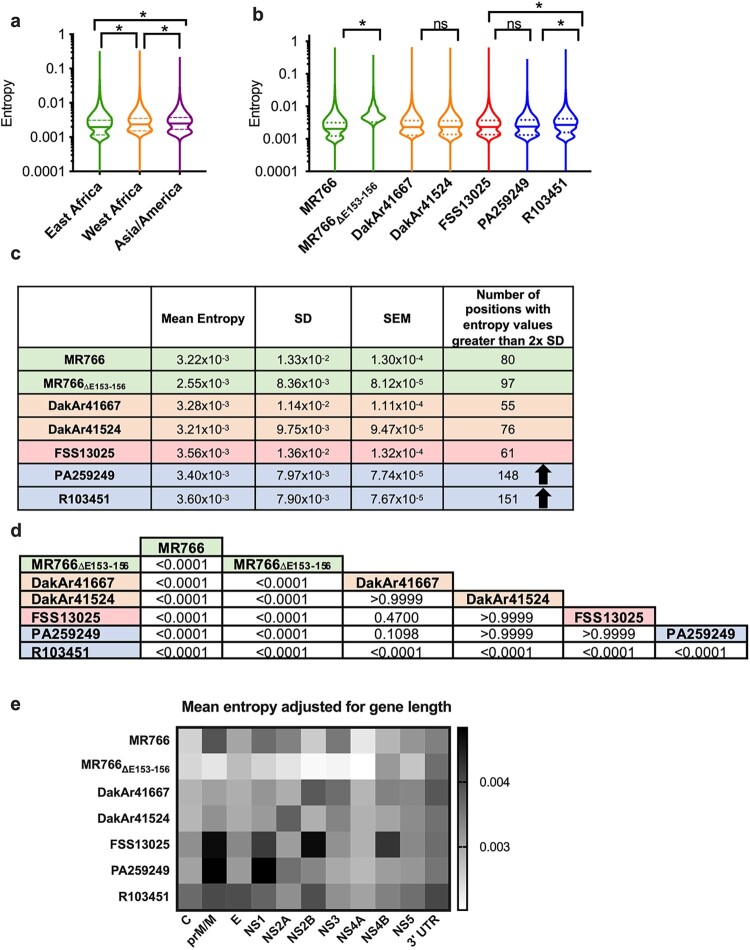
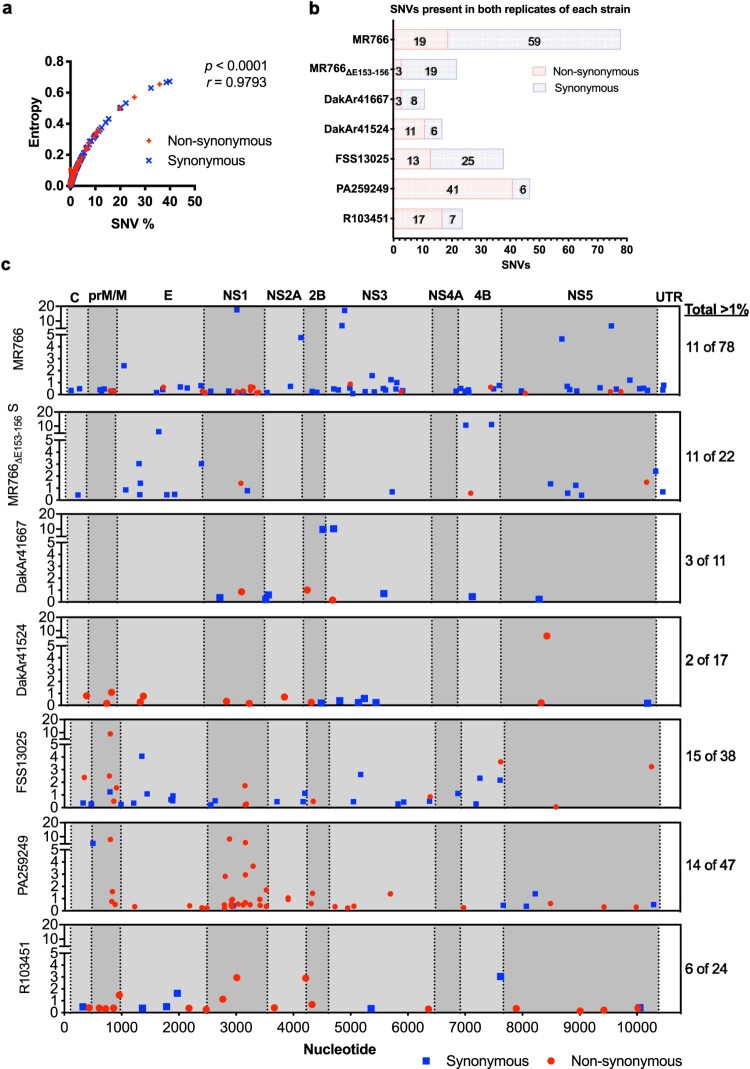
Zika virus (ZIKV) strains belong to the East African, West African, and Asian/American phylogenetic lineages. RNA viruses, like ZIKV, exist as populations of genetically-related sequences whose heterogeneity may impact viral fitness, evolution, and virulence. Genetic diversity of representative ZIKVs from each lineage was examined using next generation sequencing (NGS) paired with downstream entropy and single nucleotide variant (SNV) analysis. Comparisons showed that inter-lineage diversity was statistically supported, while intra-lineage diversity. Intra-lineage diversity was significant for East but not West Africa strains. Furthermore, intra-lineage diversity for the Asian/American lineage was not supported for human serum isolates; however, a placenta isolate differed significantly. Relative in the pre-membrane/membrane (prM/M) gene of several ZIKV strains. Additionally, the East African lineage contained a greater number of synonymous SNVs, while a greater number of non-synonymous SNVs were identified for American strains. Further, inter-lineage SNVs were dispersed throughout the genome, whereas intra-lineage non-synonymous SNVs for Asian/American strains clustered within prM/M and NS1 gene. This comprehensive analysis of ZIKV genetic diversity provides a repository of SNV positions across lineages. We posit that increased non-synonymous SNV populations and increased relative genetic diversity of the prM/M and NS1 proteins provides more evidence for their role in ZIKV virulence and fitness.
寨卡病毒(ZIKV)株属于东非、西非和亚洲/美洲进化枝。像 ZIKV 这样的 RNA 病毒以遗传相关序列的种群形式存在,其异质性可能影响病毒的适应性、进化和毒力。使用下一代测序(NGS)结合下游熵和单核苷酸变异(SNV)分析,研究了来自每个谱系的代表性 ZIKV 的遗传多样性。比较表明,谱系间的多样性具有统计学意义,而谱系内的多样性则不然。东非的谱系内多样性具有统计学意义,但西非的谱系内多样性则没有。此外,对于亚洲/美洲谱系的人血清分离株,谱系内的多样性不支持,但胎盘分离株则有显著差异。在几个 ZIKV 株的前膜/膜(prM/M)基因中也观察到了相对较高的同义 SNV 率。此外,东非谱系包含更多的同义 SNV,而美洲株的非同义 SNV 则更多。此外,谱系间的 SNV 分散在整个基因组中,而亚洲/美洲株的谱系内非同义 SNV 则集中在 prM/M 和 NS1 基因内。对 ZIKV 遗传多样性的综合分析提供了跨越谱系的 SNV 位置的存储库。我们假设,增加的非同义 SNV 群体和增加的 prM/M 和 NS1 蛋白的相对遗传多样性提供了更多证据表明它们在 ZIKV 毒力和适应性中的作用。