J. Craig Venter Institute, Rockville, MD, USA.
Sharp Edge Labs, Pittsburgh, PA, USA.
Sci Rep. 2018 Oct 26;8(1):15843. doi: 10.1038/s41598-018-34147-7.
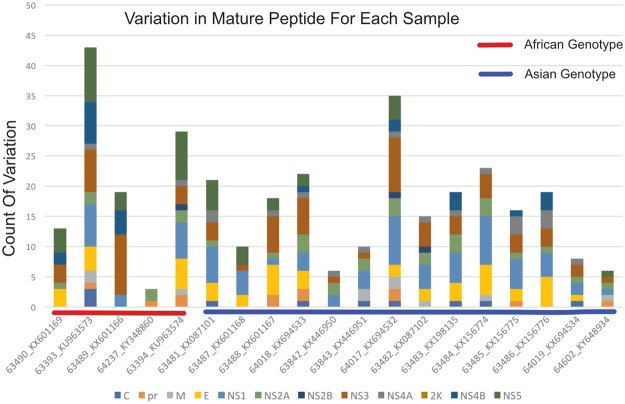
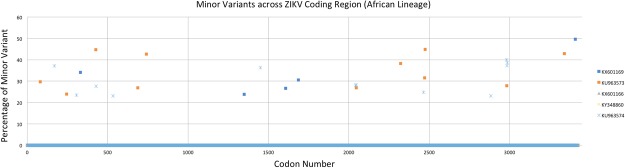
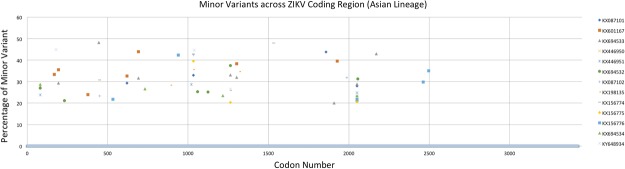
The recent emergence of Zika virus (ZIKV) has been concentrated in the Caribbean, Southeastern United States, and South- and Central America; resulting in travel-based cases being reported around the globe. As multi-disciplinary collaborations are combatting the ZIKV outbreak, the need to validate the sequence of existing strains has become apparent. Here, we report high-quality sequence data for multiple ZIKV strains made publicly available through the National Institutes of Health- (NIH) funded biorepository, BEI Resources (www.beiresources.org). Next-generation sequencing, 3' rapid amplification of cDNA ends (RACE), and viral genome annotation pipelines generated GenBank sequence records for 16 BEI Resources strains. Minor variants, consensus mutations, and consensus insertions/deletions were identified within the viral stocks using next-generation sequencing (NGS) and consensus changes were confirmed with Sanger sequencing. Bioinformatics analyses of the sequencing results confirm that the virus stocks available to the scientific research community through BEI Resources adequately represent the viral population diversity of ZIKV.
寨卡病毒(ZIKV)的近期出现主要集中在加勒比海、美国东南部以及中南美洲地区,导致世界各地都有基于旅行的病例报告。随着多学科合作对抗寨卡病毒的爆发,对现有毒株序列进行验证的需求变得明显。在这里,我们报告了通过美国国立卫生研究院(NIH)资助的生物资源库 BEI Resources(www.beiresources.org)公开提供的多个寨卡病毒株的高质量序列数据。下一代测序、3'快速扩增 cDNA 末端(RACE)和病毒基因组注释管道为 16 个 BEI Resources 株生成了 GenBank 序列记录。使用下一代测序(NGS)在病毒株中鉴定了少数变体、共识突变和共识插入/缺失,并用 Sanger 测序确认了共识变化。对测序结果的生物信息学分析证实,通过 BEI Resources 提供给科学界的病毒株充分代表了寨卡病毒病毒群体的多样性。