Institute of Computer Science, Warsaw University of Technology, ul. Nowowiejska 15/19, 00-665 Warsaw, Poland.
Gigascience. 2019 Aug 1;8(8). doi: 10.1093/gigascience/giz094.
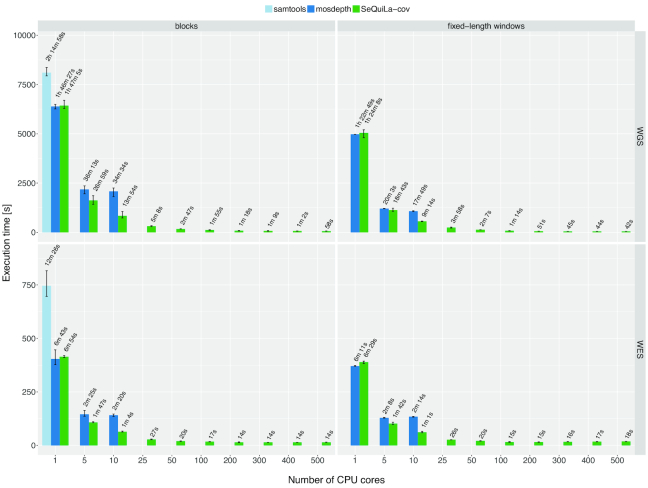
Depth of coverage calculation is an important and computationally intensive preprocessing step in a variety of next-generation sequencing pipelines, including the analysis of RNA-sequencing data, detection of copy number variants, or quality control procedures.
Building upon big data technologies, we have developed SeQuiLa-cov, an extension to the recently released SeQuiLa platform, which provides efficient depth of coverage calculations, reaching >100× speedup over the state-of-the-art tools. The performance and scalability of our solution allow for exome and genome-wide calculations running locally or on a cluster while hiding the complexity of the distributed computing with Structured Query Language Application Programming Interface.
SeQuiLa-cov provides significant performance gain in depth of coverage calculations streamlining the widely used bioinformatic processing pipelines.
在各种下一代测序管道中,覆盖深度计算是一个重要且计算密集型的预处理步骤,包括 RNA-seq 数据分析、拷贝数变异检测或质量控制程序。
基于大数据技术,我们开发了 SeQuiLa-cov,它是最近发布的 SeQuiLa 平台的扩展,提供了高效的覆盖深度计算,相对于最先进的工具实现了超过 100 倍的加速。我们的解决方案的性能和可扩展性允许在本地或集群上进行外显子组和全基因组计算,同时通过使用结构化查询语言应用程序编程接口隐藏分布式计算的复杂性。
SeQuiLa-cov 在覆盖深度计算中提供了显著的性能提升,简化了广泛使用的生物信息处理管道。