Lund University , Department of Biomedical Engineering , Lund , Sweden.
University of Groningen, Department of Analytical Biochemistry , Groningen Research Institute of Pharmacy , Antonius Deusinglaan 1 , 9713 AV Groningen , The Netherlands.
Anal Chem. 2019 Sep 17;91(18):11888-11896. doi: 10.1021/acs.analchem.9b02637. Epub 2019 Aug 23.
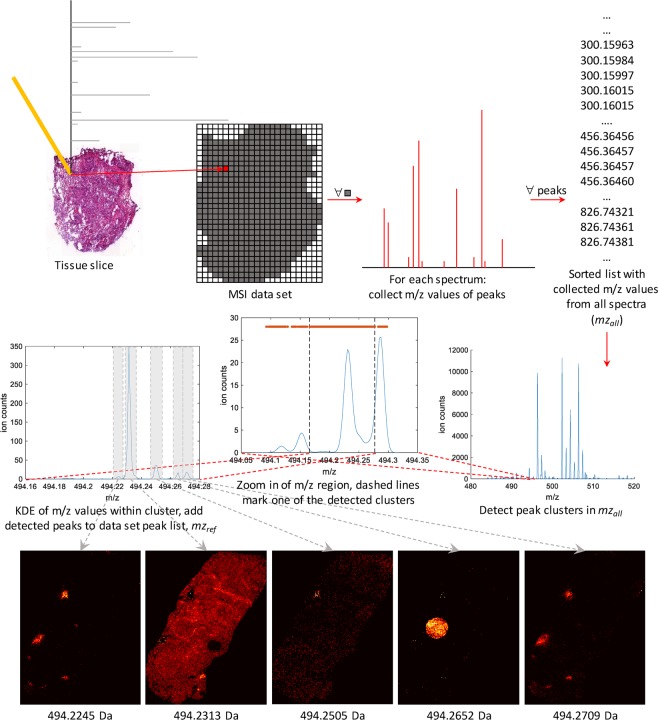
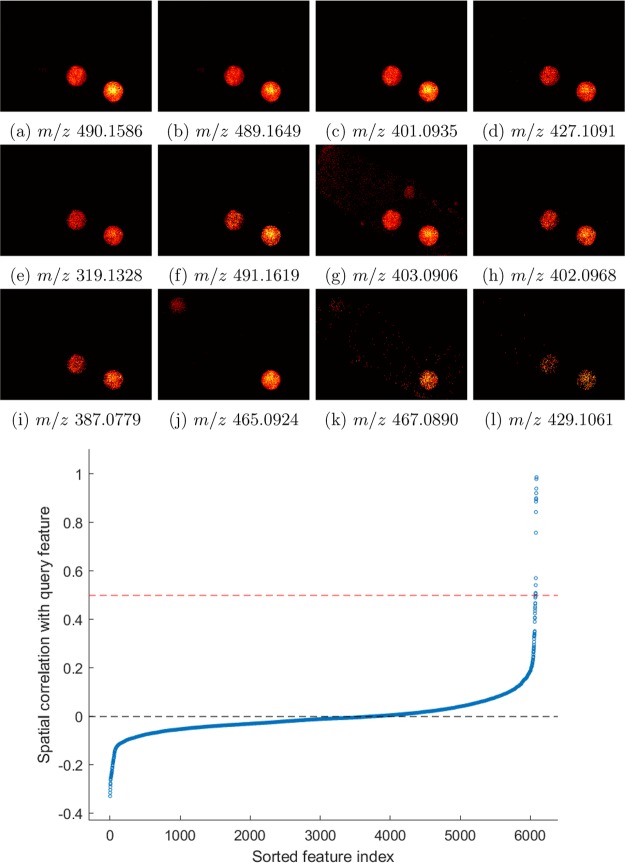
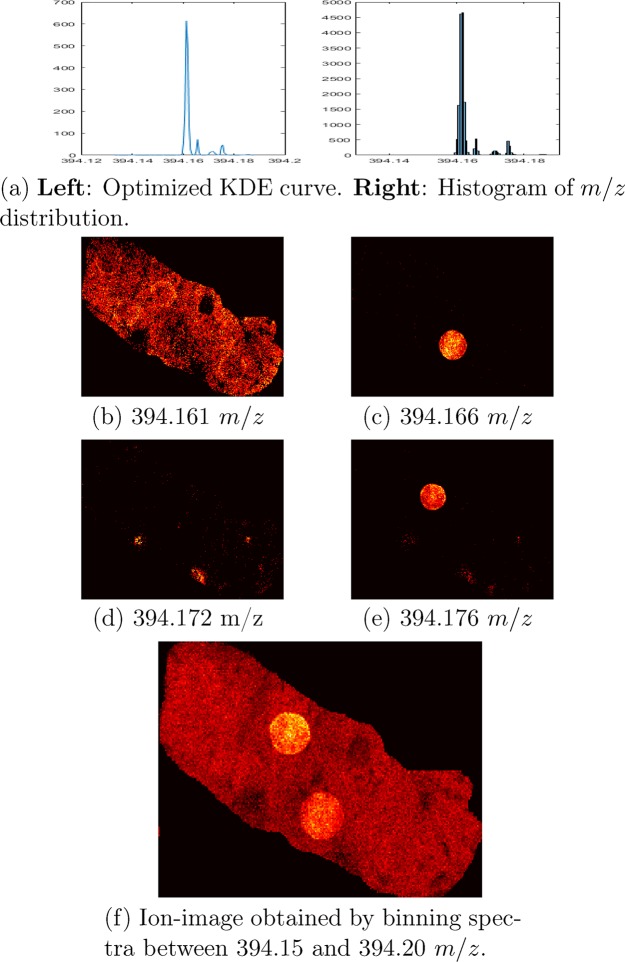
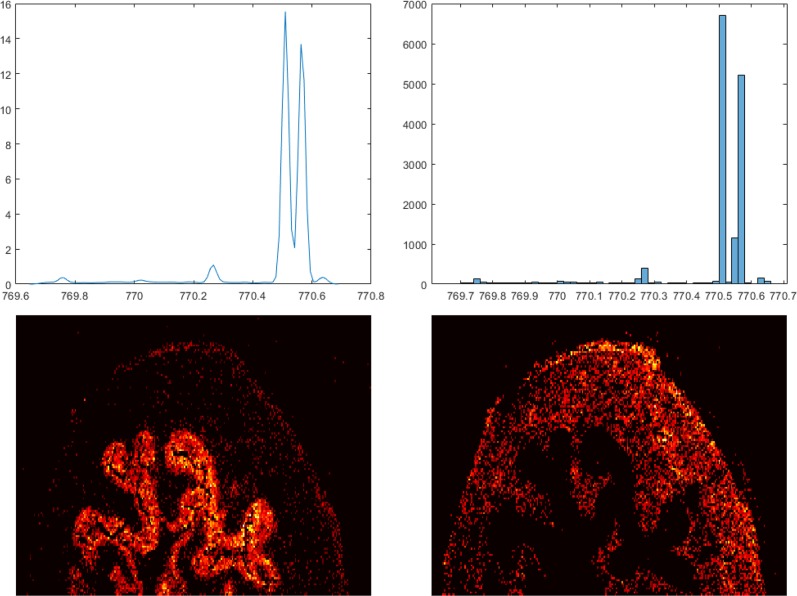
Mass spectrometry imaging (MSI) has the potential to reveal the localization of thousands of biomolecules such as metabolites and lipids in tissue sections. The increase in both mass and spatial resolution of today's instruments brings on considerable challenges in terms of data processing; accurately extracting meaningful signals from the large data sets generated by MSI without losing information that could be clinically relevant is one of the most fundamental tasks of analysis software. Ion images of the biomolecules are generated by visualizing their intensities in 2-D space using mass spectra collected across the tissue section. The intensities are often calculated by summing each compound's signal between predefined sets of borders (bins) in the / dimension. This approach, however, can result in mixed signals from different compounds in the same bin or splitting the signal from one compound between two adjacent bins, leading to low quality ion images. To remedy this problem, we propose a novel data processing approach. Our approach consists of a sensitive peak detection method able to discover both faint and localized signals by utilizing clusterwise kernel density estimates (KDEs) of peak distributions. We show that our method can recall more ground-truth molecules, molecule fragments, and isotopes than existing methods based on binning. Furthermore, it automatically detects previously reported molecular ions of lipids, including those close in /, in an experimental data set.
质谱成像(MSI)有可能揭示组织切片中数千种生物分子(如代谢物和脂质)的定位。当今仪器在质量和空间分辨率上的提高给数据处理带来了相当大的挑战;从 MSI 生成的大数据集中准确提取有意义的信号而不丢失可能具有临床相关性的信息是分析软件最基本的任务之一。通过在组织切片上采集的质谱,在 2D 空间中可视化其强度来生成生物分子的离子图像。强度通常通过在 / 维的预定义边界(箱)内对每个化合物的信号求和来计算。然而,这种方法可能会导致同一箱中不同化合物的混合信号,或者将一个化合物的信号分裂在两个相邻的箱之间,从而导致离子图像质量较低。为了解决这个问题,我们提出了一种新的数据处理方法。我们的方法包括一种敏感的峰检测方法,能够通过利用峰分布的聚类核密度估计(KDE)来发现微弱和局部化的信号。我们表明,与基于箱的现有方法相比,我们的方法可以召回更多的真实分子、分子片段和同位素。此外,它还可以在实验数据集自动检测到先前报道的脂质分子离子,包括那些在 / 附近的分子离子。