Shevtsova Elena, Vergnaud Gilles, Shevtsov Alexandr, Shustov Alexandr, Berdimuratova Kalysh, Mukanov Kasim, Syzdykov Marat, Kuznetsov Andrey, Lukhnova Larissa, Izbanova Uinkul, Filipenko Maxim, Ramankulov Yerlan
National Center for Biotechnology, Nur-Sultan, Kazakhstan.
Institute for Integrative Biology of the Cell (I2BC), CEA, CNRS, Univ. Paris-Sud, Université Paris-Saclay, Gif-sur-Yvette, France.
Front Microbiol. 2019 Aug 13;10:1897. doi: 10.3389/fmicb.2019.01897. eCollection 2019.
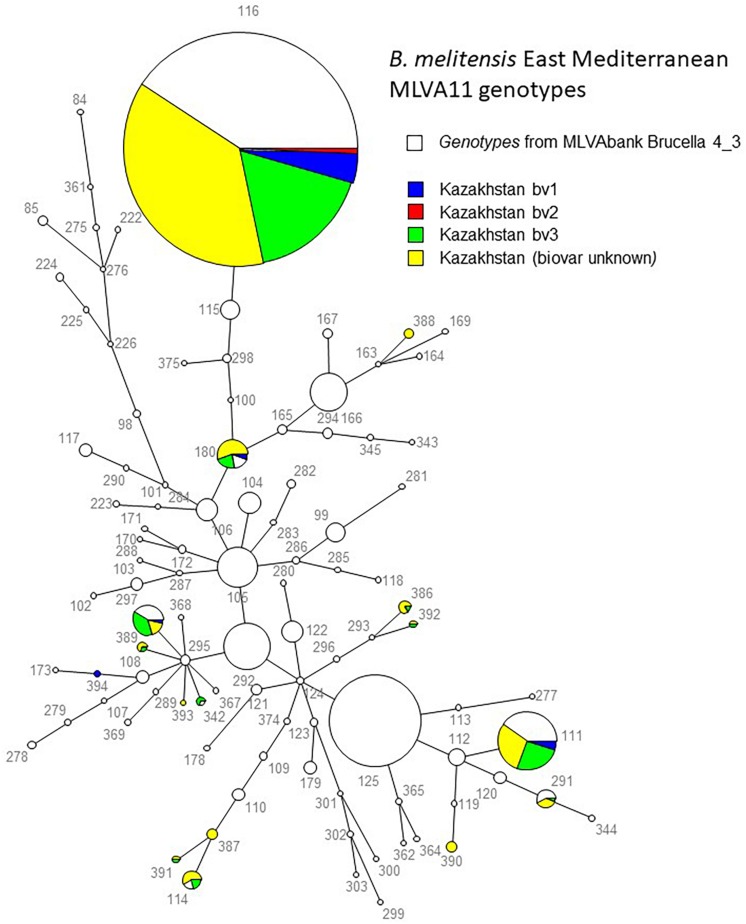
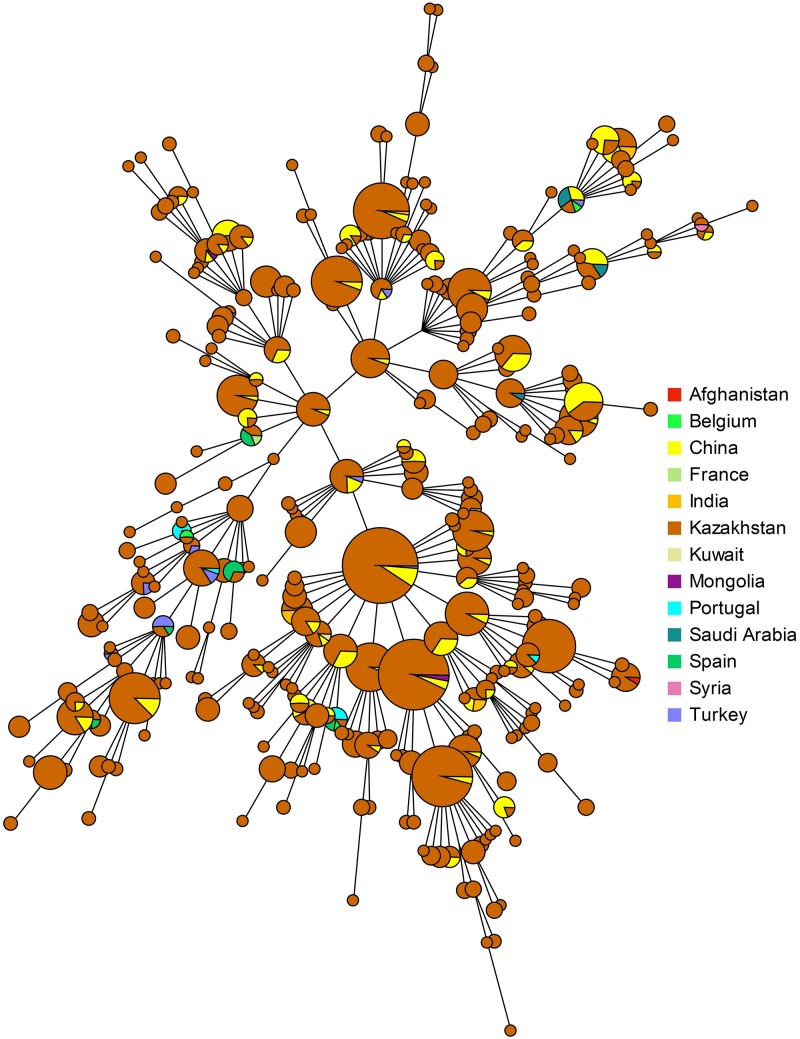
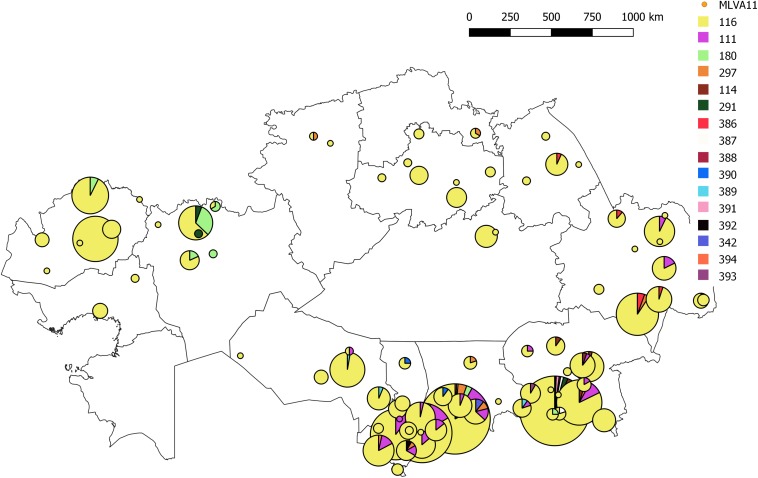
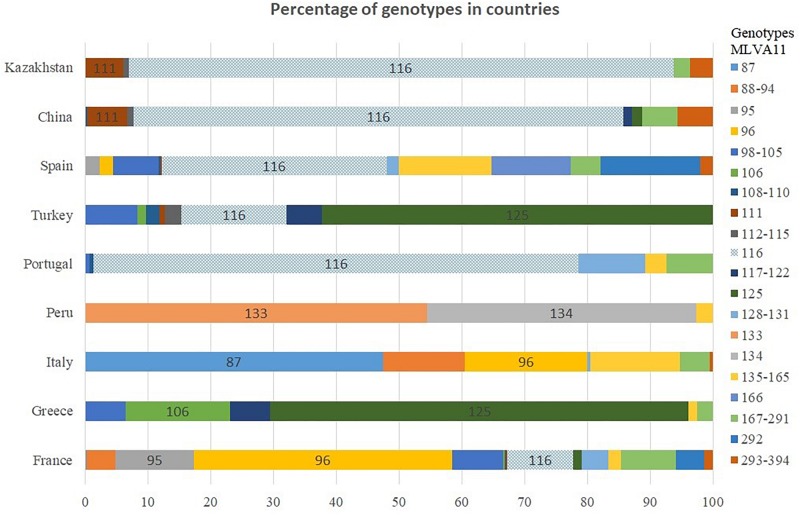
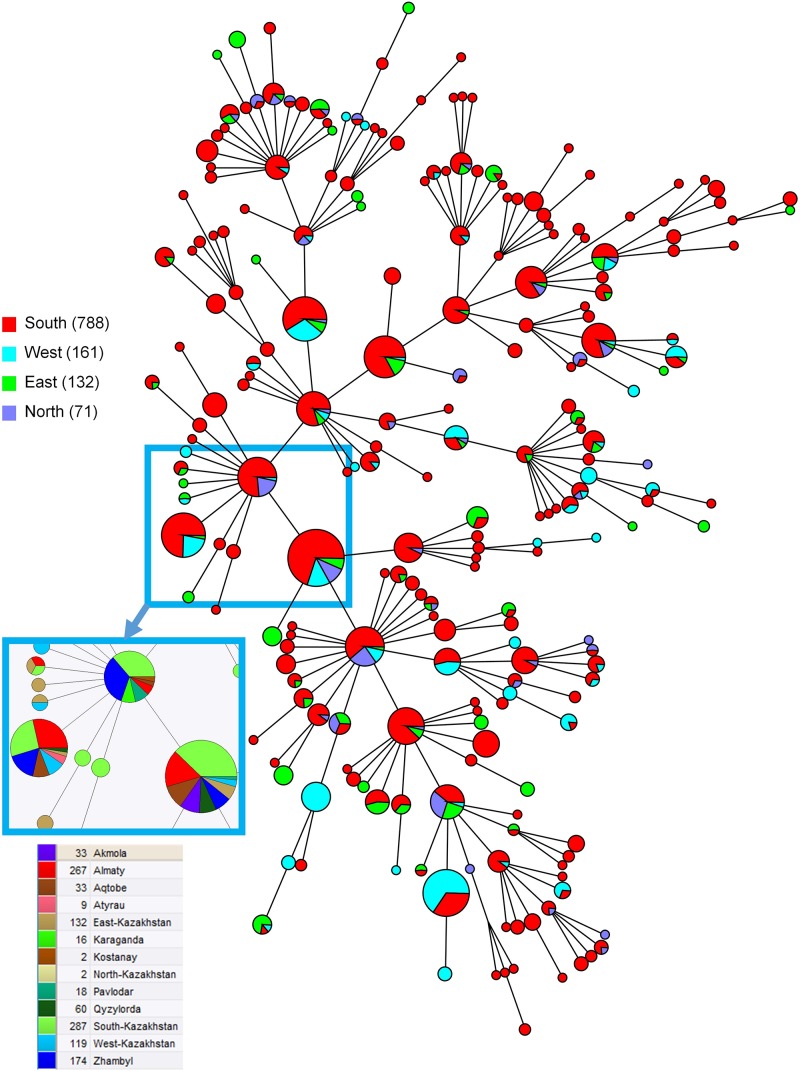
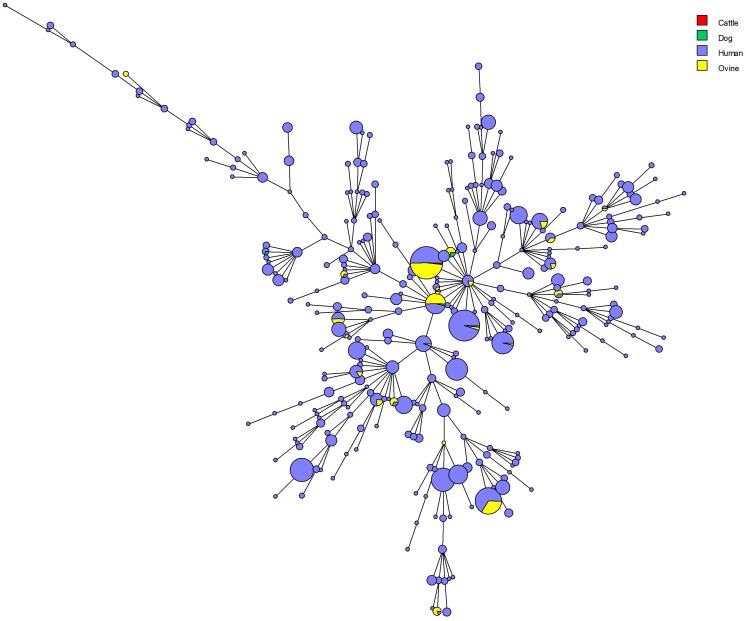
We describe the genetic diversity of 1327 strains from human patients in Kazakhstan using multiple-locus variable-number tandem repeat (VNTR) analysis (MLVA). All strains were assigned to the East Mediterranean group and clustered into 16 MLVA11 genotypes, nine of which are reported for the first time. MLVA11 genotype 116 predominates (86.8%) and is present all over Kazakhstan indicating existence and temporary preservation of a "founder effect" among strains circulating in Central Eurasia. The diversity pattern observed in humans is highly similar to the pattern previously reported in animals. The diversity observed by MLVA suggested that the epidemiological status of brucellosis in Kazakhstan is the result of the introduction of a few lineages, which have subsequently diversified at the most unstable tandem repeat loci. This investigation will allow to select the most relevant strains for testing these hypotheses via whole genome sequencing and to subsequently adjust the genotyping scheme to the Kazakhstan epidemiological situation.
我们使用多位点可变数目串联重复序列(VNTR)分析(MLVA)描述了来自哈萨克斯坦人类患者的1327株菌株的遗传多样性。所有菌株均归为东地中海组,并聚类为16种MLVA11基因型,其中9种为首次报道。MLVA11基因型116占主导地位(86.8%),在哈萨克斯坦各地均有出现,表明在中亚地区传播的菌株中存在“奠基者效应”并暂时保留。在人类中观察到的多样性模式与先前在动物中报道的模式高度相似。MLVA观察到的多样性表明,哈萨克斯坦布鲁氏菌病的流行病学状况是少数谱系引入的结果,这些谱系随后在最不稳定的串联重复位点发生了分化。这项研究将有助于选择最相关的菌株,通过全基因组测序来检验这些假设,并随后根据哈萨克斯坦的流行病学情况调整基因分型方案。