Liu Zhiguo, Wang Chengling, Wei Kongjiao, Zhao Zhongzhi, Wang Miao, Li Dan, Wang Heng, Wei Qiang, Li Zhenjun
National Institute for Communicable Disease Control and Prevention, Beijing, China.
Qinghai Institute for Endemic Diseases Prevention and Control, Xining, China.
Front Vet Sci. 2021 Jan 7;7:539444. doi: 10.3389/fvets.2020.539444. eCollection 2020.
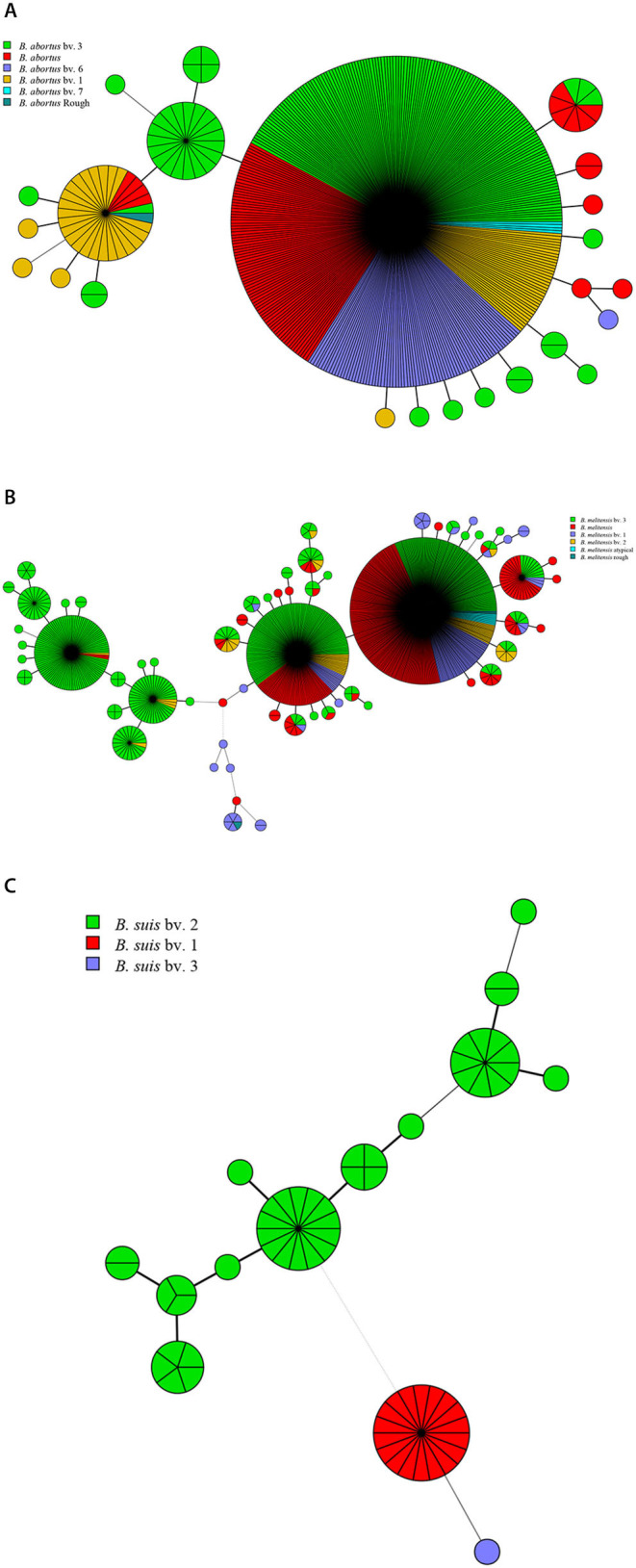
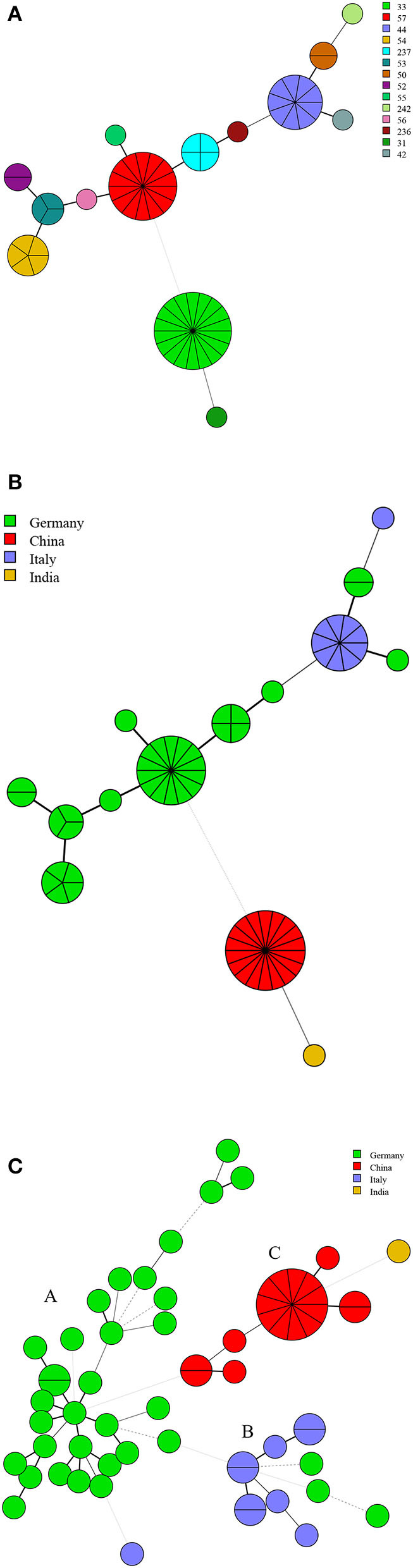
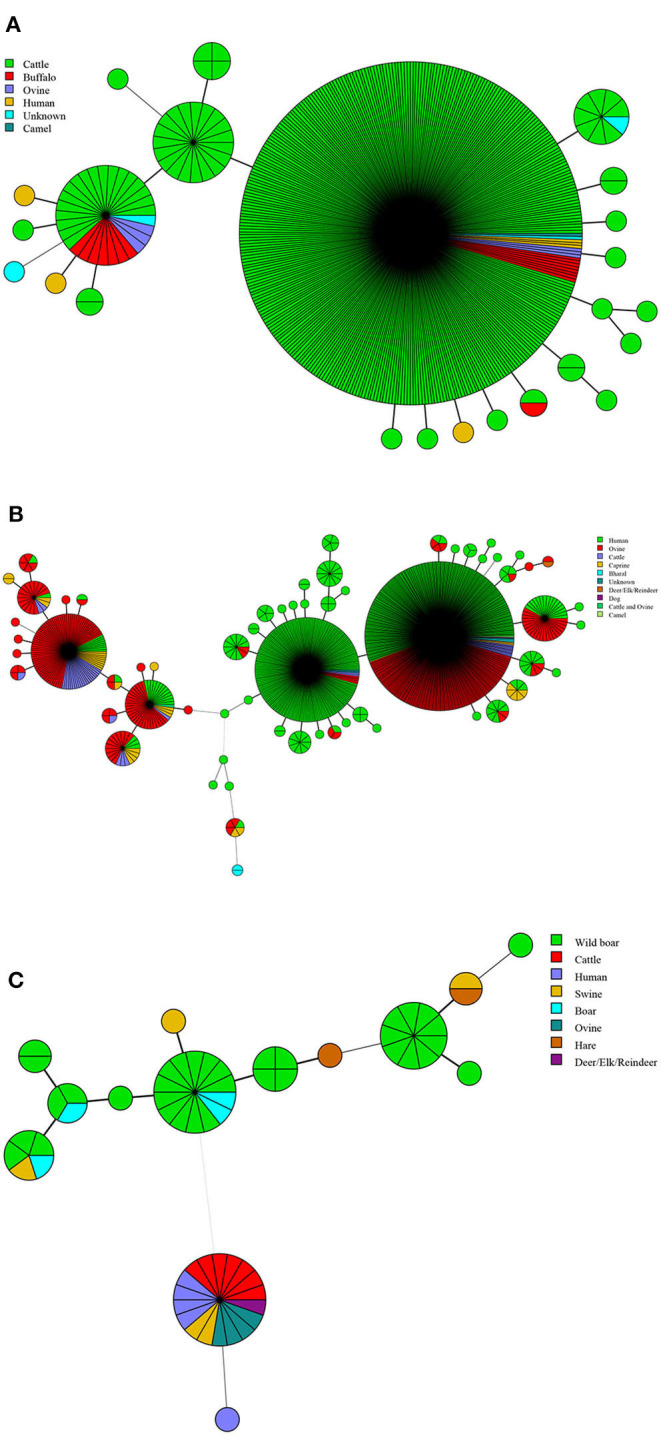
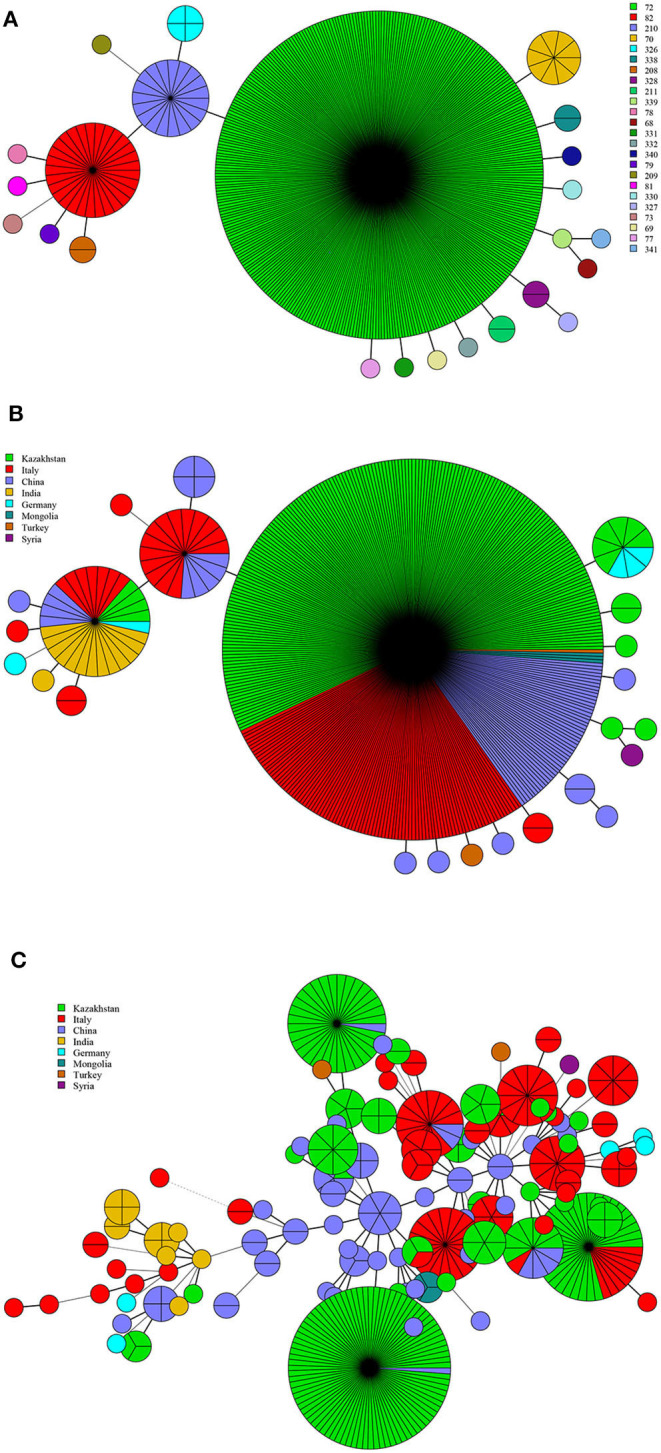
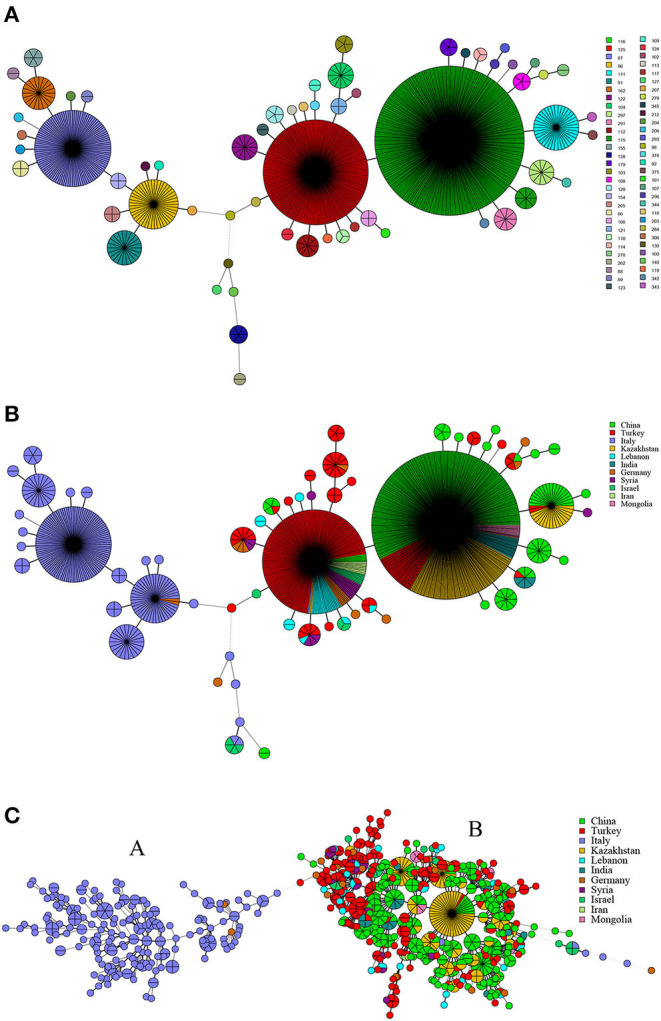
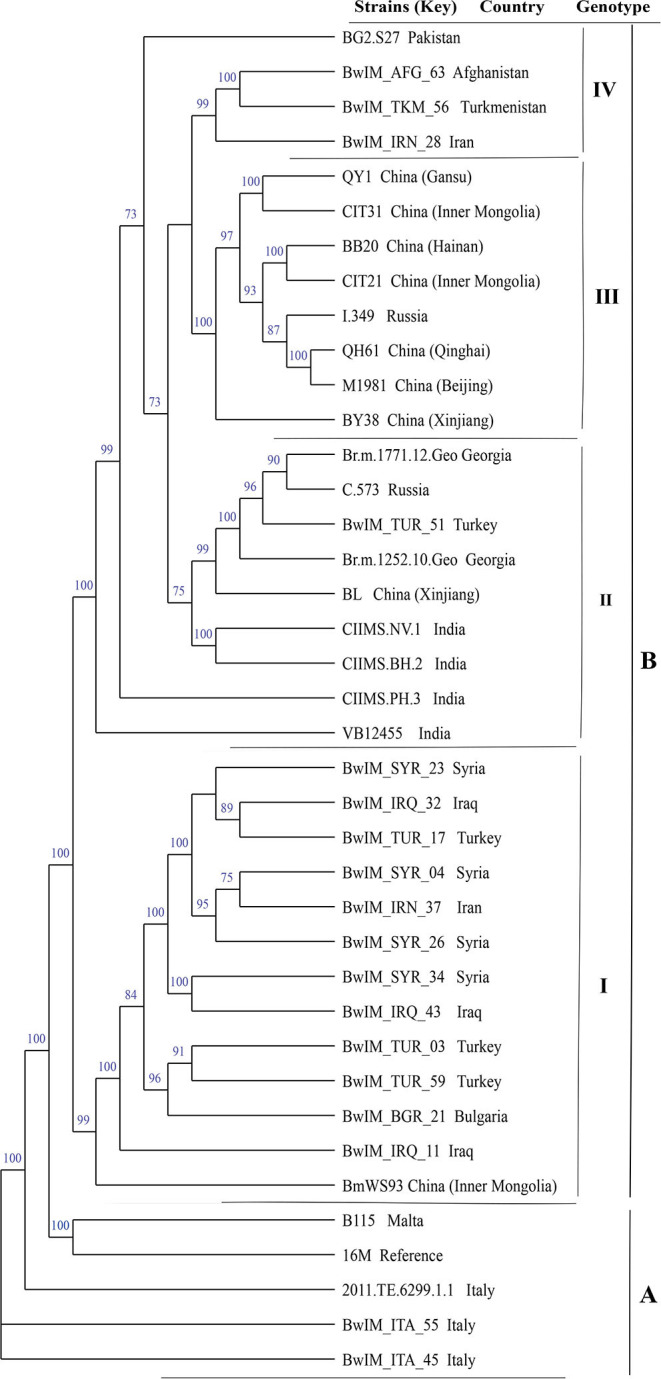
In this study, MLVA (multiple-locus variable-number tandem repeat analysis) genotype data of strains from 11 countries along the Silk Road were downloaded from the MLVAbank. MLVA data of strains were applied to the constructed Minimum Spanning Tree to explore the species/biovars distribution, geographic origins, and genetic relationships of the strains analyzed. Moreover, whole-genome sequencing-single-nucleotide polymorphism (WGS-SNP) phylogenetic analysis of the genome of strains from GenBank was performed to discriminate the relatedness of strains further and investigate the transmission pattern of brucellosis. A total of 1,503 strains were analyzed in this study: 431 strains (29.8%), 1,009 strains (65.7%), and 63 strains (4.5%). biovar 3 was the dominant species and was shown to be widespread in all of the examined regions, suggesting that the prevention and surveillance of the population are a main challenge in these countries. A wide host spectrum was observed for this population; many animal reservoirs are a potential reason for the continuous brucellosis circulation in these countries. Although the strains from the examined regions had common geographic origins, only a few shared genotypes were observed in different countries. These data revealed that the majority strains were spreading within the national borders. However, the strains from Italy originated from a Western Mediterranean lineage; strains from the other 10 countries originated from Eastern Mediterranean lineage, and this lineage was shared by strains from three to nine different countries, suggesting that the introduction and reintroduction of the disease in the 10 countries might have occurred in the past. Furthermore, the most shared MLVA-16 genotypes were formed in the strains from China, Kazakhstan, and Turkey, suggesting that the introduction and trade in sheep and goats have occurred frequently in these countries. WGS-SNP analysis showed that the in this study originated from the Malta (Italy) region. According to their territorial affiliation between four clade strains from these countries in genotype B, the absence of a clear differentiation suggests that strains continuously expand and spread in countries along with Silk Road. Active exchange and trade of animals (sheep and goats) among these countries are reasonable explanations. strains from different nations showed unique geographic origins and epidemiological characteristics. Therefore, there is an urgent need for the control of transfer and trade of infected sheep (goats) in countries along the Silk Road, namely, the strengthening of the entry-exit quarantine of sheep and goats and improvements in the diagnosis of animal brucellosis.
在本研究中,从MLVAbank下载了丝绸之路沿线11个国家菌株的多位点可变数目串联重复序列分析(MLVA)基因型数据。将菌株的MLVA数据应用于构建的最小生成树,以探索所分析菌株的种/生物变种分布、地理起源和遗传关系。此外,对GenBank中菌株的全基因组测序-单核苷酸多态性(WGS-SNP)系统发育分析进行了进一步鉴别菌株的相关性,并研究布鲁氏菌病的传播模式。本研究共分析了1503株菌株:431株(29.8%)、1009株(65.7%)和63株(4.5%)。生物变种3是优势种,在所有检测区域均广泛分布,这表明对该菌群体的预防和监测是这些国家面临的主要挑战。观察到该菌群体具有广泛的宿主谱;许多动物宿主是这些国家布鲁氏菌病持续传播的潜在原因。尽管来自检测区域的菌株有共同的地理起源,但在不同国家仅观察到少数共享基因型。这些数据表明,大多数菌株在国界内传播。然而,来自意大利的菌株起源于西地中海谱系;来自其他10个国家的菌株起源于东地中海谱系,且该谱系被三至九个不同国家的菌株共享,这表明在过去10个国家可能发生过该病的传入和再次传入。此外,中国、哈萨克斯坦和土耳其的菌株中形成了最多的共享MLVA-16基因型,这表明这些国家频繁发生绵羊和山羊的引进和贸易。WGS-SNP分析表明,本研究中的菌株起源于马耳他(意大利)地区。根据这些国家在基因型B中四个进化枝菌株之间的地域归属,缺乏明显分化表明菌株在丝绸之路沿线国家不断扩展和传播。这些国家之间活跃的动物(绵羊和山羊)交换和贸易是合理的解释。来自不同国家的菌株表现出独特的地理起源和流行病学特征。因此,迫切需要控制丝绸之路沿线国家感染绵羊(山羊)的转移和贸易,即加强绵羊和山羊的出入境检疫以及改进动物布鲁氏菌病的诊断。