Sun Mingjun, Jing Zhigang, Di Dongdong, Yan Hao, Zhang Zhicheng, Xu Quangang, Zhang Xiyue, Wang Xun, Ni Bo, Sun Xiangxiang, Yan Chengxu, Yang Zhen, Tian Lili, Li Jinping, Fan Weixing
Laboratory of Zoonoses, Chinese Animal Health and Epidemiology Center, Qingdao, China.
Xinjiang Center of Animal Disease Control, Urumqi, China.
Front Vet Sci. 2017 Dec 14;4:215. doi: 10.3389/fvets.2017.00215. eCollection 2017.
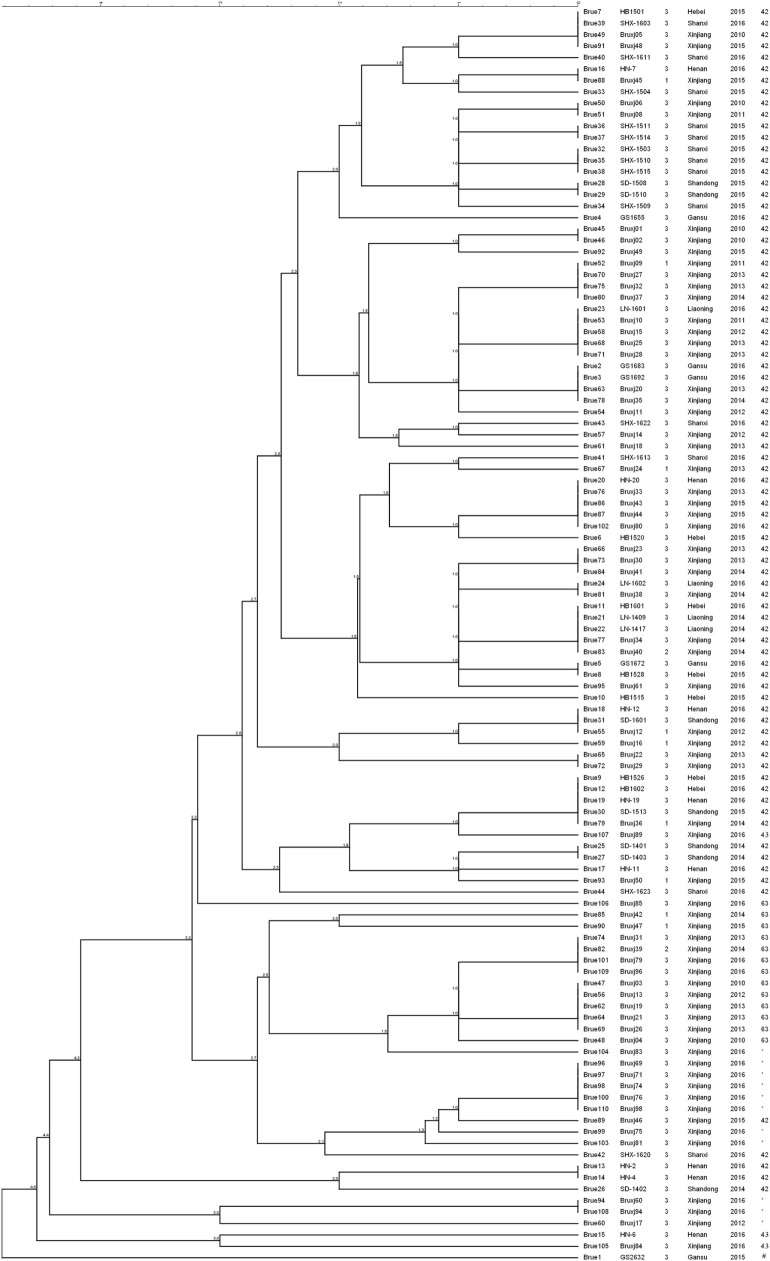
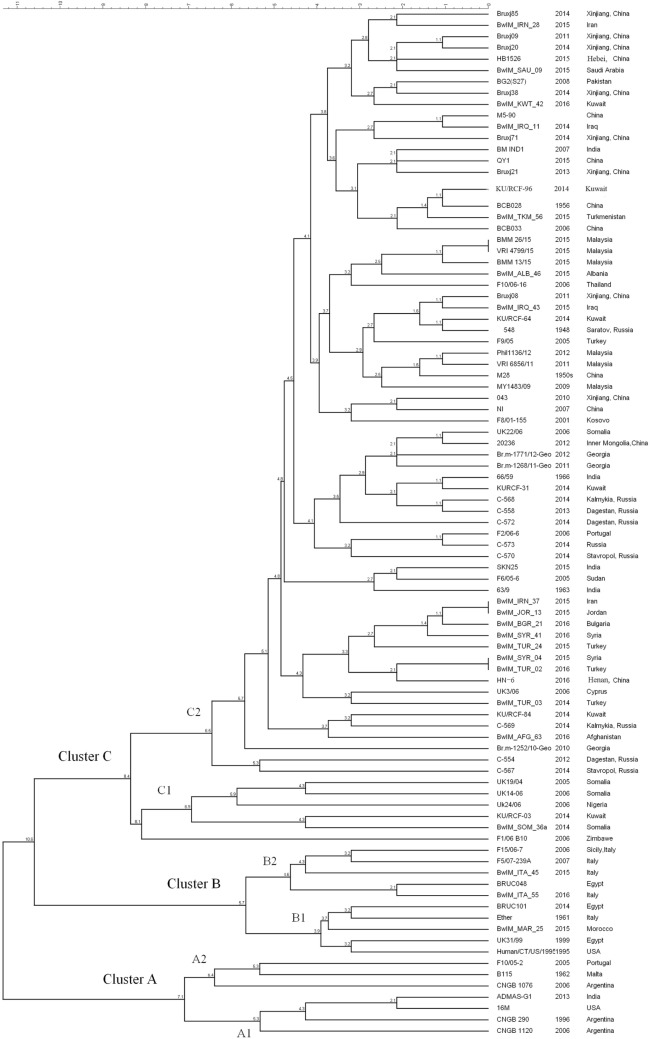
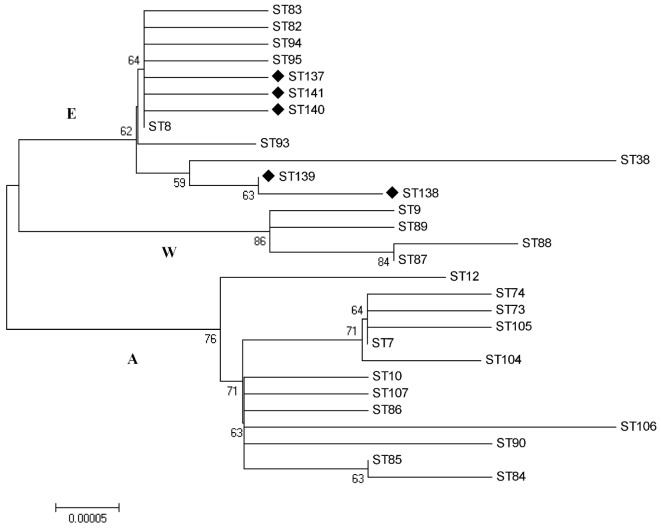
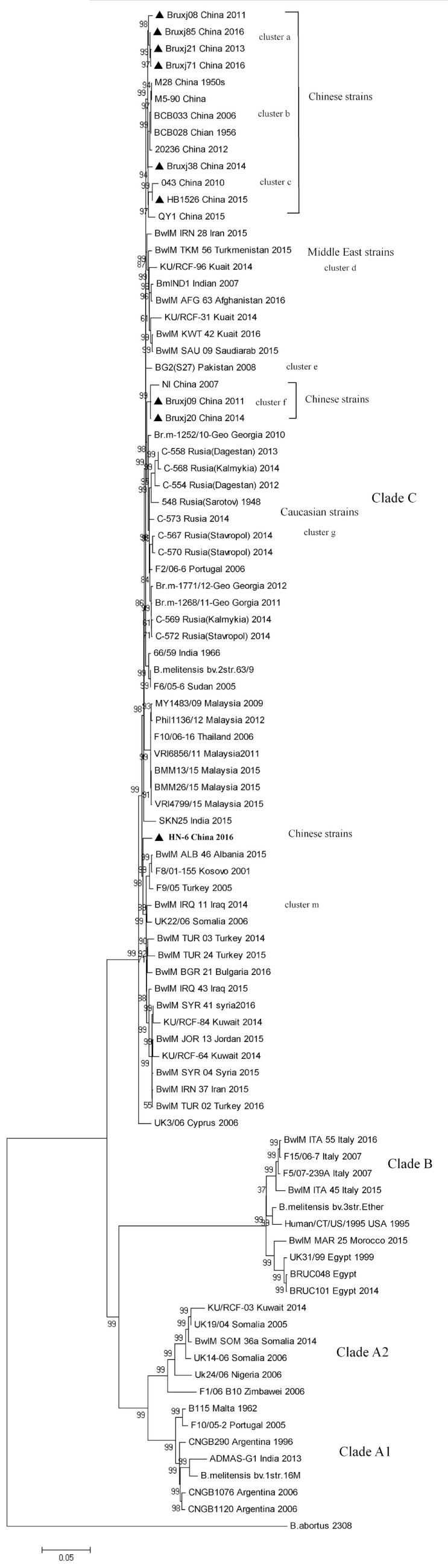
Brucellosis is a worldwide zoonotic disease caused by spp. In China, brucellosis is recognized as a reemerging disease mainly caused by specie. To better understand the currently endemic strains in China, three genotyping methods were applied to 110 strains obtained in past several years. By MLVA genotyping, five MLVA-8 genotypes were identified, among which genotypes 42 (1-5-3-13-2-2-3-2) was recognized as the predominant genotype, while genotype 63 (1-5-3-13-2-3-3-2) and a novel genotype of 1-5-3-13-2-4-3-2 were second frequently observed. MLVA-16 discerned a total of 57 MLVA-16 genotypes among these strains, with 41 genotypes being firstly detected and the other 16 genotypes being previously reported. By BruMLSA21 typing, six sequence types (STs) were identified, among them ST8 is the most frequently seen in China while the other five STs were firstly detected and designated as ST137, ST138, ST139, ST140, and ST141 by international multilocus sequence typing database. Whole-genome sequence (WGS)-single-nucleotide polymorphism (SNP)-based typing and phylogenetic analysis resolved Chinese strains into five clusters, reflecting the existence of multiple lineages among these Chinese strains. In phylogeny, Chinese lineages are more closely related to strains collected from East Mediterranean and Middle East countries, such as Turkey, Kuwait, and Iraq. In the next few years, MLVA typing will certainly remain an important epidemiological tool for infection analysis, as it displays a high discriminatory ability and achieves result largely in agreement with WGS-SNP-based typing. However, WGS-SNP-based typing is found to be the most powerful and reliable method in discerning strains and will be popular used in the future.
布鲁氏菌病是一种由布鲁氏菌属细菌引起的全球性人畜共患病。在中国,布鲁氏菌病被认为是一种主要由某一布鲁氏菌物种引起的再度出现的疾病。为了更好地了解中国目前流行的布鲁氏菌菌株,三种基因分型方法被应用于过去几年获得的110株布鲁氏菌菌株。通过多位点可变数目串联重复序列分析(MLVA)基因分型,鉴定出五种MLVA-8基因型,其中基因型42(1-5-3-13-2-2-3-2)被认为是主要基因型,而基因型63(1-5-3-13-2-3-3-2)和一种新的基因型1-5-3-13-2-4-3-2次之。MLVA-16在这些菌株中总共识别出57种MLVA-16基因型,其中41种基因型是首次检测到,另外16种基因型先前已有报道。通过布鲁氏菌多位点序列分析21(BruMLSA21)分型,鉴定出六种序列类型(STs),其中ST8在中国最为常见,而其他五种STs是首次检测到,并被国际多位点序列分型数据库指定为ST137、ST138、ST139、ST140和ST141。基于全基因组序列(WGS)-单核苷酸多态性(SNP)的分型和系统发育分析将中国布鲁氏菌菌株分为五个簇,反映了这些中国布鲁氏菌菌株中存在多个谱系。在系统发育中,中国谱系与从东地中海和中东国家收集的菌株关系更密切,如土耳其、科威特和伊拉克。在未来几年,MLVA分型肯定仍将是布鲁氏菌感染分析的重要流行病学工具,因为它具有很高的鉴别能力,并且结果在很大程度上与基于WGS-SNP的分型一致。然而,基于WGS-SNP的分型被发现是鉴别布鲁氏菌菌株最强大、最可靠的方法,并且在未来将被广泛应用。